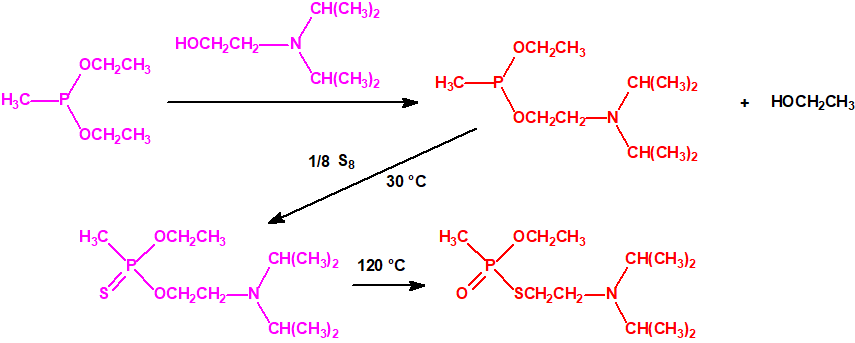
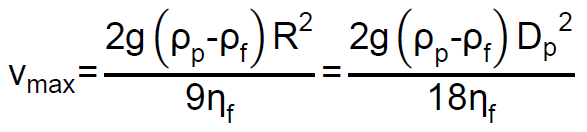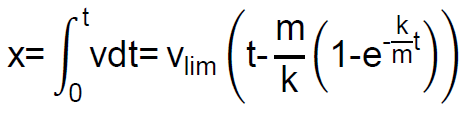Sistemas dispersos1
Se definen como sistemas dispersos aquellos que se encuentran formados por una sustancia finamente dividida y distribuida en otra. La sustancia que se distribuye se denomina dispersoide o fase interna, y la otra fase, la que recibe a la sustancia, se denomina medio de dispersión, fluido o fase externa.
Las propiedades de los sistemas dispersos varían sobre todo con el tamaño de las partículas dispersas, y según el tamaño de las mismas podríamos establecer tres categorías:
- dispersiones, tamaño de partícula superior a 100 nm (1 µm),
- coloides, tamaño comprendido entre 100 nm (1 µm) y 1 nm (0,001 µm),
- soluciones, tamaño inferior a 1 nm (0,001 µm).
Aunque las propiedades dependen fundamentalmente del tamaño de las partículas dispersas, también influye su naturaleza y la del fluido que actúa como dispersante.
Tipos de dispersiones coloidales:
|
Fase dispersa
(dispersoide o fase interna)
|
Medio de dispersión
(dispersante o fase externa)
|
Denominación
|
|
Líquido
|
Gas
|
Aerosol de gotas líquidas, niebla
|
|
Sólido
|
Gas
|
Aerosol de partículas sólidas, humo
|
|
Gas
|
Líquido
|
Espuma
|
|
Líquido
|
Líquido
|
Emulsión
|
|
Sólido
|
Líquido
|
Solución o suspensión
|
|
Gas
|
Sólido
|
Espuma sólida
|
|
Líquido
|
Sólido
|
Emulsión sólida, gel
|
|
Sólido
|
Sólido
|
Suspensión sólida, aleación
|
Se dice que un sistema disperso es estable cuando con el paso del tiempo la fase dispersa se mantiene uniformemente distribuida en la fase dispersante. La estabilidad de los sistemas depende del equilibrio de las fuerzas a las que está sometida la fase dispersa, siendo los principales fenómenos que intervienen en ello la sedimentación/cremado, la agregación y la coalescencia.
Las partículas de la fase dispersa se encuentran sometidas a las fuerzas de gravedad, empuje y rozamiento, esta última originada cuando las partículas se mueven dentro del fluido o fase dispersante. Las dos primeras actúan en dirección vertical (para la gravedad natural) y en sentidos opuestos, y provocan un movimiento en el sentido de la fuerza que predomine. Al movimiento en el seno del fluido se opone siempre la fuerza de rozamiento. Stokes estudió la acción de las fuerzas sobre partículas esféricas, llegando a determinar la velocidad máxima (velocidad límite) que pueden alcanzar, la cual viene dada por la fórmula:
donde, g es la fuerza de la gravedad, ρp es la densidad de la partícula, ρf es la densidad del fluido, Dp es el diámetro de partícula y ηf es la viscosidad del fluido
Si ρp > ρf, se produce el depósito de las partículas en el fondo del fluido, y el proceso se denomina sedimentación. Si ρp < ρf, se producirá un depósito en la parte superior del fluido, y este fenómeno se denomina flotación o cremado.
En caso de que las partículas no sean esféricas, que es lo más habitual, esto habría que tenerlo en cuenta mediante un coeficiente de forma, y además la ecuación de Stokes sólo es válida en determinadas condiciones, conocidas como «régimen laminar».
Las moléculas del fluido se mueven de forma caótica, dependiendo la velocidad, dirección y espacio recorrido de los choques entre ellas, de su concentración y de la temperatura. Cuando existen partículas pequeñas dispersas, las moléculas chocan elásticamente con ellas provocando el mismo movimiento caótico, que recibe el nombre de «movimiento browniano».
La cantidad de movimiento transferida en los choques depende de la masa de las partículas. Las grandes apenas se ven influenciadas, pero a medida que disminuye el tamaño este afecto aumenta hasta llegar a incluso a un punto en el que las fuerzas producidas por los choques prevalecen sobre las de sedimentación y las partículas se mantienen dispersas.
Si la dispersión inicial se realiza en un pequeño volumen de fluido o fase dispersante, alcanzando una determinada concentración, y el sistema disperso se pone en contacto con otro sistema disperso de menor concentración, o con la propia fase dispersante libre de fase dispersa, las partículas de la fase dispersa, debido al movimiento browniano, tienden a ocupar todo el volumen puesto a su disposición, por lo que se dispersan hasta que la concentración en todo el sistema es la misma; a este fenómeno se le denomina «difusión».
Gases, vapores y aerosoles1
Aunque los gases, los vapores y los aerosoles se comportan de manera muy similar en muchos aspectos, en otros lo hacen de manera completamente diferente.
Un gas y un vapor no son exactamente lo mismo, el vapor es un tipo de gas, pero no viceversa. Un gas es un estado de la materia. Si se comprime un gas isotérmicamente (sin cambiar la temperatura), éste nunca pasa al estado líquido a presiones elevadas, mientras que este cambio de fase sí ocurre en un vapor. Los sólidos y los líquidos pueden pasar al estado gaseoso, generando vapores, dependiendo de su punto de sublimación o de su punto de ebullición, respectivamente, aumentando la presión de vapor de los mismos. La presión de vapor es la presión que ejerce la fase gaseosa o vapor sobre la fase líquida en un sistema cerrado a una temperatura determinada, en la que la fase líquida y el vapor se encuentran en equilibrio dinámico. Su valor es independiente de las cantidades de líquido y vapor presentes mientras existan ambos. Este fenómeno también lo presentan los sólidos, de modo que cuando un sólido pasa al estado gaseoso sin pasar por el estado líquido (proceso denominado sublimación) también hablamos de presión de vapor.
Dependiendo del tipo y cantidad de gas o de vapor liberado en el aire, se puede generar o no una situación de atmósfera (aire+agente) tóxica o/y explosiva. La toxicidad depende de la concentración del agente tóxico y del tiempo de exposición a la atmósfera tóxica, mientras que las condiciones de atmósfera explosiva pueden alcanzarse entrando por el límite inferior de explosividad (LEL) al aumentar la concentración del gas, vapor o aerosol, o entrando por el límite superior de explosividad (UEL) al diluir la concentración del gas, vapor o aerosol.
Los gases, vapores y aerosoles pueden afectar nuestra salud por inhalación o contacto, pero la inhalación es con mucho la vía principal y la más peligrosa.
Los aerosoles son dispersiones coloidales en las que se dispersa un sólido o un líquido en una fase gaseosa continua. Se distinguen dos tipos principales de aerosoles en la ciencia coloidal y en la nanotecnología:
- aerosoles sólidos (aerosoles de partículas sólidas), para partículas sólidas dispersas en un gas, y
- aerosoles líquidos (aerosoles de gotitas de líquido), para gotitas de líquido dispersas en un gas.
Frecuentemente se utiliza el término aerosol para referirse a productos empleados en forma de aerosol mediante su liberación de un envase a presión a través de un fino orificio que produce un aerosol o una espuma.
Las partículas de un aerosol pueden ser tan pequeñas como 1 nm (0,001 µm) y las gotas de un aerosol pueden ser tan grandes como aproximadamente 100 μm.
Los aerosoles de partículas sólidas o gotitas de líquido a veces se distinguen por su mecanismo general de creación:
- aerosoles primarios, los creados por la dispersión de partículas, agregados o gotas (polvo, neblina), o mediante reacciones químicas (hollín), y emitidos desde una fuente, o
- aerosoles secundarios, los creados en la atmósfera por condensación de gases con formación de sólidos o líquidos (por ejemplo, humo, vapores, neblina).
En general, las partículas gruesas de un aerosol, como, por ejemplo, el polvo del suelo, las gotas de las nubes y las partículas biológicas, se producen mediante procesos mecánicos. Las partículas o gotitas de un aerosol formadas a partir de la fase gaseosa suelen tener un tamaño más pequeño y, a menudo, tienen un diámetro inferior a 1 µm. Estas partículas finas provienen generalmente de fuentes de emisión industriales o se forman en la atmósfera.
En la práctica, los aerosoles tienen rangos de tamaño que van desde grupos moleculares en la nanoescala (1 nm y mayores), hasta polvos y nubes que contienen gotitas de aerosol que exceden los límites del rango clásico de tamaño coloidal y que pueden alcanzar hasta los 100 µm.
En los temas referentes a los aerosoles se distinguen frecuentemente los siguientes tipos:
- polvo: aerosoles de partículas sólidas (de aproximadamente más de 0,5 µm de diámetro) resultado de la desintegración mecánica de partículas más grandes.
- humos: aerosoles de partículas sólidas (de menos de 1 µm de diámetro) que surgen de la condensación de los vapores de una reacción química o física (como la evaporación y la condensación).
- niebla: aerosoles de gotitas de líquido. En algunas definiciones, la niebla se caracteriza por un rango de tamaño de gota particular, mientras que, en otras, la niebla se refiere a la niebla que tiene una concentración de gotas lo suficientemente alta como para oscurecer la visibilidad.
- niebla con humo (smog): aerosoles de gotitas líquidas o partículas sólidas que comprenden contaminación del aire (con diámetros inferiores a aproximadamente 2 µm).
- humo: aerosoles de gotitas líquidas o partículas sólidas que resultan de procesos térmicos como combustión o descomposición térmica.
- bioaerosoles: partículas en el aire que son de origen biológico, como células bacterianas dispersas y esporas de hongos, fragmentos de insectos u otros animales y partículas portadoras de virus.
El comportamiento y la trayectoria de los aerosoles se ven fuertemente afectados por la naturaleza, tamaño y aerodinámica de las partículas, sus interacciones y los efectos provocados por las condiciones meteorológicas. Conocer el comportamiento de los aerosoles es de vital importancia tanto en la generación de los mismos para aprovechar sus beneficios, como para defendernos de sus inconvenientes.
Las partículas de los aerosoles están influenciadas por su tamaño, su densidad, su naturaleza, el viento, la temperatura, la humedad relativa, etc., pudiendo disminuir o aumentar su tamaño, flotar o sedimentar, permaneciendo más o menos tiempo suspendidas en el aire.
Sedimentación de los aerosoles2,3
Una partícula esférica inmersa en un fluido se mueve bajo la acción de las siguientes fuerzas: el peso, el empuje (se supone que el cuerpo está completamente sumergido en el seno de un fluido), y una fuerza de rozamiento que es proporcional a la velocidad de la partícula esférica (suponemos que el flujo se mantiene en régimen laminar).
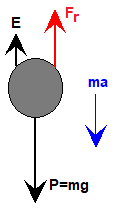
El peso «P» de la partícula esférica es el producto de su masa por la aceleración de la gravedad «g», y puesto que su masa es el producto del volumen de una esfera de radio R por la densidad de la partícula esférica ρp:
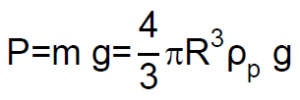
Por otro lado, de acuerdo con el principio de Arquímedes, el empuje «E» es igual al producto del volumen del cuerpo sumergido por la densidad del fluido «ρf«, y por la aceleración de la gravedad «g»:
La fuerza de rozamiento «Fr» que experimenta un cuerpo moviéndose en un fluido es proporcional a su velocidad «v», y su expresión se denomina ley de Stokes:
donde ηf es la viscosidad del fluido.
La ecuación del movimiento será, por tanto:
La velocidad límite, se alcanza cuando la aceleración «a» es cero, es decir:
Despejamos la velocidad límite vlim:
Como puede apreciarse la velocidad límite para una partícula suspendida en el aire es directamente proporcional a su densidad y proporcional al cuadrado de su tamaño.
Puesto que la ecuación del movimiento para la esfera sumergida en un fluido es:
donde F es la diferencia entre el peso y el empuje, F=P-E, y k=6πRη
Integramos la ecuación del movimiento para obtener la velocidad de la partícula esférica en función del tiempo:
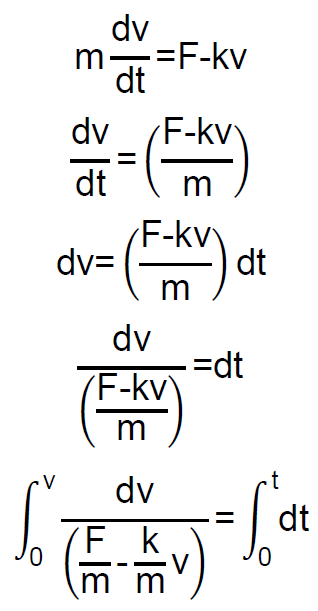
Obtenemos:

Y como:
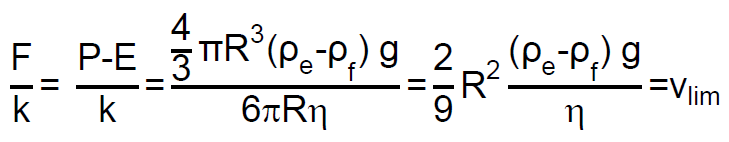
Tendremos:

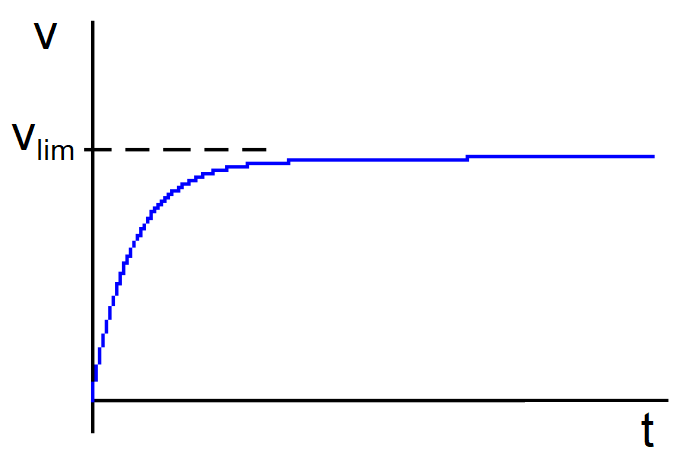
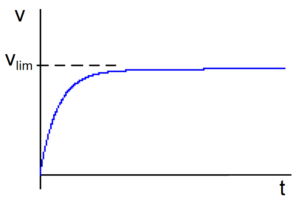
Esta ecuación nos dice que se alcanza la velocidad límite vlim después de un tiempo teóricamente infinito. Si representamos v en función del tiempo t la gráfica tienen una asíntota horizontal en v=vlim.
Como 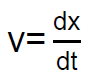 , una nueva integración nos da:
, una nueva integración nos da:
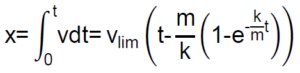
Dado que la exponencial tiende a cero rápidamente a medida que transcurre el tiempo, vemos que, al cabo de un cierto tiempo, el espacio recorrido «x» por la partícula esférica será proporcional al tiempo t.
Las diferencias entre el movimiento de un cuerpo en caída libre (sin flotación ni resistencia a la velocidad) y cuando cae en el seno de un fluido viscoso (con flotación y resistencia a la velocidad) se pueden resumir en el siguiente cuadro:
|
Caída libre
|
En el seno de un fluido viscoso
|
- La velocidad es proporcional al tiempo
|
- La velocidad tiende hacia un valor constante
|
- El espacio recorrido es proporcional al cuadrado del tiempo
|
- El espacio recorrido es proporcional al tiempo.
|
Cuanto más densas y más grandes sean las partículas más fácil y rápidamente sedimentarán las mismas y menos afectadas serán por las condiciones meteorológicas. Las partículas pequeñas y de poca densidad flotan con facilidad, están más tiempo suspendidas y el viento puede desplazarlas grandes distancias (e incluso evaporarlas) antes de que se depositen.
En ausencia de otras fuerzas y fenómenos que puedan alterar el tamaño de las partículas de un aerosol, la velocidad límite de sedimentación en el aire aumenta al aumentar la densidad y al aumentar el tamaño de la partícula. La tabla siguiente muestra la velocidad límite y la sedimentación en 1 hora, para una partícula esférica en función de su densidad:
|
Diámetro
|
ρ=1,00 g/cm3 (Agua, VX)
|
ρ=1,24 g/cm3 (Iperita)
|
ρ=1,89 g/cm3 (Lewisita)
|
ρ=18,95 g/cm3 (Uranio)
|
|
0,5 µm
|
0,000777 cm/s
2,80 cm
|
0,000963 cm/s
3,47 cm
|
0,001469 cm/s
5,29 cm
|
0,014738 cm/s
53,06 cm
|
|
1 µm
|
0,003107 cm/s
11,19 cm
|
0,003854 cm/s
13,87 cm
|
0,005876 cm/s
21,15 cm
|
0,058952 cm/s
212,2 cm
|
|
5 µm
|
0,077677 cm/s
279,6 cm
|
0,096344 cm/s
346,8 cm
|
0,146900 cm/s
528,8 cm
|
1,473789 cm/s
53,06 m
|
|
10 µm
|
0,310710 cm/s
11,19 m
|
0,385376 cm/s
13,87 m
|
0,587599 cm/s
21,15 m
|
5,895154 cm/s
212,2 m
|
|
50 µm
|
7,767744 cm/s
279,6 m
|
9,634411cm/s
346,8 m
|
14,68997 cm/s
528,8 m
|
147,3789 cm/s
5,306 km
|
|
100 µm
|
31,07098 cm/s
1,119 km
|
38,53764 cm/s
1,387 km
|
58,75987 cm/s
2,115 km
|
589,5154 cm/s
21,22 km
|
|
200 µm
|
124,2839 cm/s
4,474 km
|
154,1506 cm/s
5,549 km
|
235,0395 cm/s
8,461 km
|
2358,062 cm/s
84,89 km
|
Sedimentación, evaporación y deriva4
En muchos casos interesa conocer cómo sedimenta un aerosol, y si lo hace en la zona de interés o sufre una «deriva». Por ejemplo, cuando nuestro interés es generar aerosoles para dispersar una sustancia, la deriva es un aspecto a controlar, que está influenciado por muchos factores entre los que podemos citar las características de la dispersión, el equipo y las técnicas de dispersión, así como por las condiciones atmosféricas
La deriva se asocia con el movimiento físico de las partículas del aerosol que provoca su sedimentación fuera de la vertical del punto de formación del aerosol. Este tipo de deriva se denomina «deriva por viento» y depende de la naturaleza y tamaño de las partículas, y de las condiciones meteorológicas del momento. Los aerosoles con partículas de gran tamaño presentan muy poca «deriva por viento», pero los aerosoles con partículas pequeñas pueden viajar cientos de metros antes de depositarse en el suelo. Además, en el caso de aerosoles con partículas líquidas, estas se evaporan tanto más rápido cuanto más pequeñas son, debido a su mayor superficie de evaporación, y conforme van evaporándose su tamaño va haciéndose menor.
A veces también se habla de otro tipo de deriva, una vez producida la sedimentación del aerosol. Recibe el nombre de «deriva por vapor», al asociarse con la volatilización (cambio de estado de líquido a gas) de las gotas sedimentadas del aerosol. La deriva por vapor solo es importante si la volatilidad de la sustancia líquida del aerosol es muy elevada y las condiciones atmosféricas son favorables para la vaporización.
Entre las características más importantes de la dispersión podemos citar el tamaño de las partículas, y la composición química y evaporación del material a dispersar (las partículas).
El tamaño de las partículas es con mucho el factor más importante que afecta a la deriva. Las partículas grandes con tamaños de 150 µm a 200 µm presentan una deriva insignificante para velocidades del viento entre 1,5 km/hora y 14 km/hora. En los aerosoles con tamaños de partícula inferiores a 50 µm de diámetro, las partículas permanecen suspendidas en el aire mucho tiempo hasta que se evaporan o son capturadas. En la mayor parte de los casos para reducir la deriva interesa un balance apropiado entre gotas grandes, que presentan una baja deriva, y gotas pequeñas, que proporcionan una buena cobertura. El tamaño de gotas recomendada para la dispersión de fungicidas, insecticidas y herbicidas es de 150-400 µm.
La composición química del material a dispersar afecta a su viscosidad, y esta viscosidad afecta al tamaño de las gotas. La mayor viscosidad aumenta el tamaño de las gotas de modo que hay menos gotas pequeñas que son las que más deriva sufren. Pueden añadirse aditivos al producto para incrementar su viscosidad, y entre otras cosas, aumentar el tamaño de las gotas.
La evaporación se refiere a la cantidad de partículas que pasan del estado líquido al estado gaseoso durante la vida del aerosol. La evaporación es más fácil en un aerosol de partículas pequeñas debido a que existe una mayor superficie de contacto con el aire. El fenómeno de evaporación es esencialmente un proceso de transferencia de calor y de transferencia de masa (una parte o la totalidad de la masa de las partículas se transfiere al aire en forma de vapor) como consecuencia de las diferencias de temperatura entre las partículas y el aire circundante que las rodea, y por efecto de la humedad relativa del aire.
La evaporación de las partículas afecta al tamaño de las mismas, y el tamaño de éstas afecta a su evaporación. Para un mismo volumen total, cuanto más pequeñas sean las gotas, mayor será la superficie total de evaporación y más fácil y rápidamente se evaporarán y se irán haciendo más pequeñas. Las gotas de menos de 30 µm acaban por evaporarse antes de sedimentar, mientras que las gotas de más de 150 µm no experimentaran una reducción significativa de tamaño antes de sedimentar.
Si consideramos las partículas esféricas su volumen viene dado por V=(4/3)πR3 y su superficie por S=4πR2. A partir del volumen (0,004189 mm3) de una gota de tamaño 200 µm (de radio 100 µm) se podrían generar 106 (un millón) de gotas 2µm de tamaño (cada una con un volumen de 4,189×10-9 mm3). Puesto que la superficie de una gota de tamaño 200 µ es de 0,12566 mm2 y la de una gota de tamaño 2 µm es de 0,12566×10-4 mm2, la superficie de 1 millón de gotas de tamaño 2 µm es cien veces la superficie de una gota de tamaño 200 µm. La evaporación es un fenómeno superficial, y puesto que las gotas más pequeñas suponen mayor superficie, las gotas pequeñas se evaporarán más rápido que las gotas grandes.
En general, podemos afirmar que la evaporación será mayor en los aerosoles con partículas acuosas pequeñas, con temperaturas ambientales altas y/o humedades relativas del aire. Igualmente, habrá mayor evaporación cuanto mayor sea el tiempo que permanecen suspendidas las partículas, expuestas a las condiciones meteorológicas. Como se mencionó anteriormente, durante el proceso de evaporación las gotas transfieren masa al aire circundante, disminuyendo su tamaño. En ciertos casos la evaporación es un factor crítico, pues además aumenta el riesgo de deriva5.
El proceso de generación de gotas para conseguir un espray o aerosol se llama atomización o nebulización, y suele llevarse a cabo haciendo pasar un líquido a través de una boquilla. El tipo de boquilla condiciona la forma o patrón del espray, y el tamaño de sus partículas, de modo que el tamaño de partícula aumenta al elegir una boquilla de tipo pulverización fina, cono hueco, abanico plano y cono lleno. Las condiciones de operación también influyen en el tamaño de las partículas: las presiones más altas producen partículas más pequeñas y las presiones más bajas producen partículas más grandes, las boquillas de flujo más bajo producen partículas más pequeñas y las boquillas de flujo más alto producen partículas más grandes, un aumento de la tensión superficial del líquido aumentan el tamaño de partícula, las partículas pequeñas pueden tener una velocidad inicial más alta, pero la velocidad disminuye rápidamente, mientras que las partículas más grandes retienen la velocidad por más tiempo y viajan más lejos6,7.
Las condiciones atmosféricas pueden afectar de manera importante el comportamiento de los aerosoles. Varios factores asociados al microclima del lugar pueden afectar al tamaño de las partículas, a su sedimentación y evaporación, y a la deriva del aerosol. Estos factores incluyen la velocidad y dirección del viento, la humedad relativa y la temperatura, y la estabilidad atmosférica e inversión térmica.
Resulta obvio que, a mayor velocidad del viento, más lejos puede ser transportada una gota de un determinado tamaño. La dirección del viento es importante para reducir el daño causado por la deriva.
La humedad relativa y la temperatura no son tan críticas como la velocidad del viento, pero tienen su importancia en algunas áreas geográficas o bajo ciertas condiciones meteorológicas. A medida que una partícula cae a través del aire, las moléculas de agua de la superficie de la gota se evaporan. Esta evaporación reduce el tamaño y la masa de la gota, pudiendo así permanecer en el aire durante más tiempo y, bajo las condiciones adecuadas, desplazarse mayores distancias. Aunque las pérdidas por evaporación en los aerosoles ocurren bajo casi todas las condiciones atmosféricas, estas pérdidas son menos importantes durante los momentos más fríos del día como al amanecer o al atardecer. Además, la humedad relativa es usualmente mayor durante estos momentos fríos.
La estabilidad atmosférica influencia en forma significativa la deriva. Bajo condiciones meteorológicas normales, la temperatura del aire decrece 1 ºC cada 120 metros de altura. El aire frío tiende a asentarse, desplazando el aire caliente de abajo y causando un mezclado vertical. Mientras que las capas de aire caliente ascienden, las gotas suspendidas suben con él y se disipan dentro de las capas superiores por la natural turbulencia y mezcla vertical del aire. Sin embargo, pueden surgir problemas cuando la atmósfera es muy estable. Bajo condiciones de estabilidad, una capa de aire caliente ubicada arriba a cierta distancia puede constituirse en una manta, manteniendo abajo el aire frío. Este fenómeno es conocido como inversión atmosférica. Las partículas suspendidas en la capa fría no pueden moverse para ningún lado excepto lateralmente, posiblemente por algunos kilómetros. Eventualmente, la suspensión puede encontrar una corriente de aire descendente, forzando las partículas a caer y depositándose fuera del objetivo, quizá sobre un cultivo sensible. Nuevamente, la mejor manera de evitar la deriva asociada a la inversión atmosférica es evitar la formación de pequeñas partículas (150 µm o menores) durante la aspersión.
Inhalación de aerosoles8,9
El aire que respiramos no contiene únicamente nitrógeno y oxígeno. Se considera que, el aire es una mezcla de gases en proporciones ligeramente variables, compuesto por un 21% de oxígeno, un 78% de nitrógeno, un 0,93 % de argón, 0,04 % de dióxido de carbono y pequeñas cantidades de otros gases, así como una cantidad variable de vapor de agua (alrededor de un 1 % al nivel del mar y de un 0,4 % en toda la atmósfera). El aire que respiramos contiene, además, ingentes cantidades de partículas en suspensión, tanto sólidas como líquidas, orgánicas e inorgánicas, bacterias, virus, pólenes, polvo, y otras partículas más o menos simples.
El aparato respiratorio está formado por las vías aéreas y por los pulmones. A través de las vías aéreas el aire circula en dirección a los pulmones y es en estos órganos donde se realiza el intercambio de gases. En las vías aéreas diferenciamos la vía aérea superior, que va desde la nariz y la boca hasta las cuerdas vocales, e incluye la faringe y la laringe, y la vía aérea inferior, formada por la tráquea, los bronquios y sus ramificaciones en el interior de los pulmones, los bronquiolos.
El sistema respiratorio está especialmente diseñado, tanto anatómica como funcionalmente, para que el aire llegue a los territorios más distales en las mejores condiciones de limpieza. En la vía aérea superior, los pelos de la nariz, las fosas nasales, las cuerdas vocales, los cilios del epitelio bronquial, los reflejos del estornudo y de la tos, etc., contribuyen a realizar esta limpieza.
En la vía aérea inferior, el árbol bronquial comienza en la tráquea, y en la carina o bifurcación traqueal tiene lugar la primera bifurcación de la tráquea en los dos bronquios principales, uno relacionado con cada pulmón. El bronquio principal derecho es más corto, ancho y verticalizado que el izquierdo. Los bronquios principales se dividen dicotómicamente en bronquios lobares, los lobares se dividen y forman los bronquios segmentarios. Sucesivamente esto últimos se dividen en subsegmentarios grandes, luego subsegmentarios pequeños, bronquios terminales, hasta llegar a lo que se conoce como acino respiratorio, el cual se forma por el bronquiolo respiratorio, los sacos alveolares, y los alveolos. En total tenemos aproximadamente 23 generaciones (bifurcaciones) en el árbol bronquial hasta llegar a los alveolos10.
A medida que aumenta el número de generaciones (es decir, a medida que las vías respiratorias se hacen más pequeñas), la cantidad de cilios, la cantidad de células secretoras de moco, la presencia de glándulas submucosas y la cantidad de cartílago en las paredes de las vías respiratorias disminuyen gradualmente. El moco es importante para atrapar las partículas pequeñas. Los cilios barren la alfombra de moco, que se mantiene húmeda por las secreciones de las glándulas submucosas, hacia la faringe, donde al tragar se elimina el moco. El cartílago es importante para prevenir el colapso de las vías respiratorias, que es especialmente un problema durante la espiración. Las vías respiratorias mantienen algo de cartílago hasta aproximadamente la décima generación, hasta el momento en que se denominan bronquios11.
A partir de aproximadamente la undécima generación, las vías respiratorias ahora libres de cartílago se denominan bronquiolos. Debido a que carecen de cartílago, los bronquiolos pueden mantener abierto el paso solo porque la presión que los rodea puede ser más negativa que la presión interna y debido al tirón hacia afuera (tracción radial o inmovilización) de los tejidos circundantes. Por tanto, los bronquiolos son especialmente susceptibles al colapso durante la espiración. Hasta la generación ∼16, no hay alvéolos, y el aire no puede intercambiarse con la sangre capilar pulmonar11.
El tamaño y forma de las partículas, la velocidad del aire respirado, la geometría de las vías aéreas, el grado de humedad y los mecanismos de aclaramiento son factores que afectan al depósito de las partículas de los aerosoles.
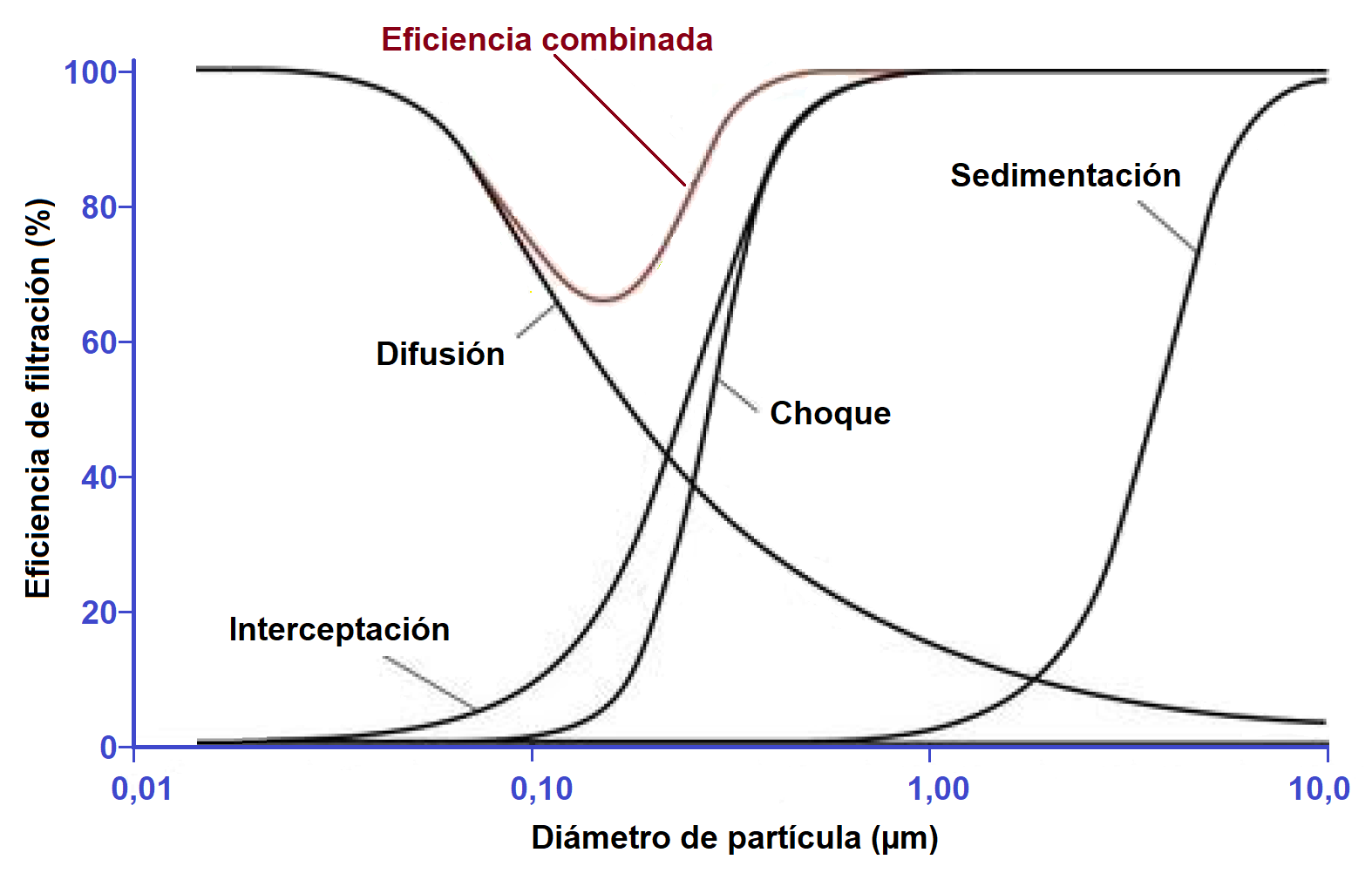
- El tamaño y la forma de las partículas son factores primordiales que van a condicionar su captura por los pulmones. El tamaño se define mediante lo que se denomina diámetro de la masa media aerodinámica (DMMA) o diámetro de una partícula de masa igual a la mediana de las partículas de una población, es decir, aquel diámetro de la partícula en el que el 50% de la masa del aerosol se encuentra por encima del mismo y el otro 50% por debajo En función de su tamaño y de su forma, las partículas pueden ser capturadas mediante uno o más de los siguientes mecanismos:
- Choque
- Interceptación
- Sedimentación
- Difusión
Choque es el fenómeno físico por el que las partículas de un aerosol tienden a continuar con su trayectoria cuando discurren por la vía aérea, en vez de adecuarse a las curvaturas del tracto respiratorio. Las partículas que tengan suficiente momento (producto de la masa por la velocidad) se verán afectadas por las fuerzas centrífugas en aquellos puntos en que el flujo de aire cambie de dirección repentinamente, chocando contra la pared de la vía aérea. Esto sucede principalmente en las primeras 10 generaciones bronquiales, en las que la velocidad del aire es elevada y el flujo es turbulento. Este fenómeno afecta sobre todo a las partículas mayores de 10 µm, que van a quedar retenidas principalmente en la región orofaríngea. Este mecanismo explica el comportamiento de las partículas más grandes en el aire, que, en lugar de seguir las líneas de flujo del aire, siguen, debido a su inercia, una trayectoria recta, impactan con los obstáculos y resultan capturadas
La interceptación (no la intercepción) se describe como un mecanismo de captura para partículas grandes. Estas partículas grandes, aunque fluyen con las líneas de flujo del aire, sobresalen tanto de las mismas que contactan con los obstáculos cuando pasan junto a ellos y resultan capturadas. Se da principalmente en el caso de las partículas fibrosas por su forma alargada.
La sedimentación es el fenómeno físico por el que las partículas con una masa suficiente se depositan por acción de la gravedad cuando el tiempo de permanencia en la vía aérea es suficientemente largo. Predomina en las 5 últimas generaciones bronquiales, en las que la velocidad del aire es baja y, por lo tanto, el tiempo de residencia se prolonga.
La difusión (no la suspensión) es el fenómeno por el que las partículas de un aerosol de desplazan de forma errática de un sitio a otro de las vías aéreas. Sucede como consecuencia del movimiento browniano de las partículas y se da en aquellas de tamaño inferior a 0,5 µm de DMMA cuando alcanzan los espacios alveolares, en donde la velocidad del aire es prácticamente nula. Estas partículas por lo general no llegan a depositarse y son expulsadas nuevamente al exterior con la espiración. Las partículas pequeñas y ligeras son capturadas por difusión. Debido a su pequeño tamaño son amortiguadas por las propias partículas del aire y tienen por ello un movimiento aleatorio que aumenta la probabilidad de que entren en contacto y sean capturadas.
De modo general puede considerarse que las partículas con DMMA mayor de 10 µm se depositan en la orofaringe, las de 5-10 µm en las vías aéreas centrales, y las de 0,5-5 µm en las pequeñas vías aéreas y alvéolos. Por lo tanto, para el tratamiento respiratorio tópico interesa emplear partículas con DMMA comprendido entre 0,5 y 5 µm. Es lo que se denomina fracción respirable de un aerosol.
- La velocidad del aire varía a lo largo de las vías respiratorias. Dado que las partículas son transportadas en la vía aérea por una corriente de aire, sus trayectorias se van a ver afectadas por las características de dicha corriente. El flujo de aire en los pulmones está determinado por el volumen corriente (cantidad de aire que entra en los pulmones con cada inspiración normal. Su valor normal es de 500 ml aproximadamente) y la frecuencia respiratoria (número de ciclos respiratorios que ocurren por minuto, es decir, número de inspiraciones seguidas de una espiración, que se pueden contar en un minuto. Lo habitual es que esté en torno a 12-16 respiraciones por minuto). Parece demostrado que en las 4 primeras generaciones de la vía aérea el depósito de partículas aumenta según lo hace el flujo inspiratorio, para cualquier tamaño de partícula. Sin embargo, lo contrario sucede en las últimas generaciones de la vía aérea, en donde el depósito de partículas es inversamente proporcional a este flujo. Esto es debido a que el incremento del flujo inspiratorio disminuye el tiempo de permanencia de las partículas en la vía aérea, por lo que los efectos de la gravedad y del movimiento browniano se verían muy reducidos.
- La geometría de las vías aéreas afecta al depósito de partículas en las mismas. Las probabilidades de depósito de las partículas por choque aumentan cuanto mayor es el tamaño de las propias partículas, cuanto mayor sea el flujo de aire inspirado, cuanto mayor sea el ángulo de separación entre dos ramas y cuanto más estrecha sea la vía aérea. La disminución del diámetro interior de la vía aérea aumenta la velocidad del aire, produciendo turbulencia en lugares en los que el flujo es normalmente laminar. La obstrucción de la vía aérea también hace que el aire tienda a desplazarse a zonas sin obstruir, por lo que las partículas del aerosol tenderán a depositarse mayoritariamente en las zonas sin obstruir (sanas) de los pulmones.
- En cuanto al grado de humedad, las partículas de los aerosoles pueden ser higroscópicas en mayor o menor medida. La higroscopicidad es la propiedad de algunas sustancias de absorber y exhalar la humedad según el medio en que se encuentran. Esto hace que puedan aumentar o disminuir de tamaño al penetrar en la vía aérea, con la consiguiente modificación del patrón de captura respecto al esperado inicialmente. El diámetro que alcanza una partícula después de su crecimiento higroscópico depende de su diámetro inicial, de las propiedades intrínsecas de la partícula y de las condiciones ambientales de las vías aéreas. En general se considera que el crecimiento higroscópico afecta poco a las partículas con DMMA inferior a 0,1 µm, mientras que es muy importante en las partículas con DMMA superior a 0,5 µm.
- El sistema mucociliar está formado por el epitelio ciliar, que tapiza la vía aérea desde la nariz hasta los bronquiolos, y por el moco, secretado por las células caliciformes y las células submucosas que se encuentran en el epitelio de la vía aérea, que generan una delgada capa de moco que recubre los cilios. Las partículas una vez depositadas en las vías aéreas, pueden ser arrastradas por el sistema mucociliar, y degradadas o absorbidas en la circulación sistémica o en los conductos linfáticos. Las partículas que alcanzan a depositarse en los alvéolos pueden ser fagocitadas y eliminadas por los macrófagos alveolares, en el caso de que sean partículas, o bien ser absorbidas hacia la circulación sistémica si son solubles.
Los aerosoles en la guerra química
La munición química se diseña:
- bien para generar un aerosol con un tamaño de partícula adecuado (de 1µm a 7µm), que permanezca suspendido en aire en una zona próxima al suelo (1-3 metros) para que pueda ser inhalado, o
- bien para generar una nube de partículas más gruesas que se deposite sobre el terreno provocando su contaminación.
En ambos casos con el fin de ocasionar bajas al enemigo y/o conseguir una disminución de sus capacidades operativas al requerir el empleo de medios de protección.
La persistencia puede definirse como el tiempo durante el cual un agente químico, en atmósfera libre y en su punto de dispersión, actúa conservando el grado de eficacia establecido. La persistencia puede variar desde algunas horas a varias semanas, en función de:
- la naturaleza del agente
- el método de dispersión
- el terreno y las condiciones meteorológicas
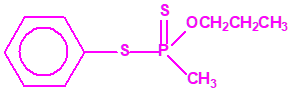
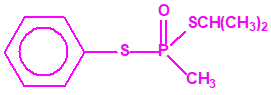
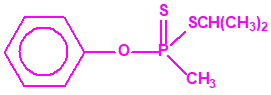
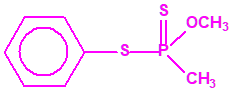
Aunque se han espesado numerosos agentes químicos de guerra, iperita, lewisita, tabún, sarín, VX, etc., el agente espesado preferentemente es el somán (GD). Las formas espesadas de somán, se suelen identificar con las siglas TGD, del inglés “Thickened GD”.
Los espesantes se añaden a los agentes químicos para aumentar su viscosidad. Uno de los espesantes más empleados es un copolímero, no tóxico, mezcla de acrilato de butilo-acrilato de etilo-metacrilato de metilo, con el nombre comercial de «Acryloid K125», que se añade en una proporción del 5%.
Al aumentar la viscosidad del agente químico:
- Se reduce la superficie total de las gotitas de agente (porque éstas se extienden menos), disminuye la evaporación y el agente resulta más persistente.
- Las gotas se adhieren mejor a las superficies y dificultan su remoción por métodos físicos.
- Aumenta la exactitud de ataque, debido a que las gotitas que se forman, son de mayor tamaño y sedimentan más rápidamente, evitándose que floten a la deriva.
- Se disminuye su extensión sobre una superficie, y su penetración en los materiales porosos se realiza más lentamente.
- Las heridas contaminadas con agentes espesados requieren mayores precauciones (Nunca debe olvidarse el peligro que conllevan los vapores del agente químico de guerra).
Aerosoles y transmisión de enfermedades (COVID-19)
Después de casi un año de pandemia y al igual que sucedió con las mascarillas, ahora parece que, con la evidencia científica acumulada, el virus SARS-CoV-2 «puede transmitirse de persona a persona por diferentes vías, siendo la principal mediante el contacto y la inhalación de las gotas y aerosoles respiratorios emitidos por un enfermo hasta las vías respiratorias superiores e inferiores de una persona susceptible»12. También se puede producir el contagio por contacto indirecto a través de las manos u objetos contaminados las secreciones respiratorias del enfermo con las mucosas de las vías respiratorias y la conjuntiva de la persona susceptible.
Todas las personas, al hablar y respirar emiten, a partir de sus vías respiratorias, aerosoles de diferentes tamaños que oscilan desde nanómetros hasta cientos de micrómetros. Según los tamaños de estos aerosoles, el comportamiento aerodinámico es diferente. Se considera que tan sólo las secreciones superiores a 100 µm tienen comportamiento «balístico» depositándose en pocos segundos por efecto de la gravedad y pueden recorrer una distancia máxima de dos metros desde el emisor. Estas emisiones podrían alcanzar a una persona susceptible que estuviera cerca impactando en algún lugar (ojos, boca, nariz) desde el cual podría causar la infección. Cualquier otra emisión respiratoria menor de 100 µm se considera un aerosol, puesto que queda suspendido en el aire por un tiempo (desde segundos hasta horas) en el que puede ser inhalado a una distancia superior a dos metros del emisor o incluso en ausencia de un emisor, si aún persisten partículas suspendidas en el aire. En función de su tamaño, los aerosoles desde 15 µm hasta 100 µm alcanzan las vías respiratorias superiores, los aerosoles desde 5 µm hasta 15 µm pueden alcanzar la tráquea y bronquios principales y los aerosoles menores o iguales a 5 µm tienen capacidad para llegar hasta los alveolos12.
A la vista de las evidencias encontradas hasta la fecha, se pueden establecer que:
- Los aerosoles generados contienen virus
- Los virus contenidos en los aerosoles tienen capacidad de generar infección sobre todo en determinadas circunstancias: en proximidad al caso índice durante tiempo prolongado y en espacios cerrados y mal En estas condiciones pueden coexistir varios mecanismos de transmisión.
- Los tejidos diana son accesibles, para aerosoles de cualquier tamaño con puertas de entrada en cualquier lugar del tracto respiratorio
Por todo lo anterior se concluye que en el estado actual del conocimiento científico existen evidencias científicas consistentes que permiten afirmar que la transmisión del virus SARS-CoV-2 por aerosoles debe considerarse como la principal vía de transmisión. Estos aerosoles podrían tanto impactar y depositarse en las conjuntivas y la mucosa del tracto respiratorio superior, como ser inhalados llegando a cualquier tramo del tracto respiratorio. El riesgo de esta transmisión aumenta en la distancia corta, en entornos cerrados y concurridos, especialmente mal ventilados, y si se realizan actividades que aumenten la generación de aerosoles como hacer ejercicio físico, hablar alto, gritar o cantar12.
La Organización Mundial de la Salud, a fecha 20 de octubre de 2020, indicaba que el virus SARS-CoV-2 se puede propagar a través de pequeñas partículas líquidas expulsadas por una persona infectada a través de la boca o la nariz al toser, estornudar, hablar, cantar o resoplar, y que esas partículas líquidas tienen diferentes tamaños, desde las más grandes «gotículas respiratorias» hasta las más pequeñas, llamadas «aerosoles». Añadía además que «la transmisión por aerosoles puede producirse en entornos específicos, sobre todo en espacios interiores, abarrotados y mal ventilados en los que personas infectadas pasan mucho tiempo con otras, por ejemplo, restaurantes, prácticas de coro, clases de gimnasia, clubes nocturnos, oficinas y/o lugares de culto.»13
Lo cierto es que, aunque el mecanismo de transmisión exacto del SARS-CoV-2 sigue sin estar claro14,15,16, se acepta, en general que la vía aérea es la principal ruta de transmisión17. Los virus respiratorios, incluido el SARS-CoV-2, se pueden dispersar a través de gotitas expulsadas por una persona infectada al toser, estornudar, hablar e incluso respirar18.
Tocarse la cara es un mecanismo potencial de transmisión secundaria del SARS-CoV-2, y la inhalación directa de gotitas cargadas de virus o núcleos de gotitas es otro14,15,19,20.
Varios estudios han informado acerca de las distribuciones de tamaño, en términos de su tamaño, de las gotas generadas a través de actividades espiratorias14,21,22,23,24,25,26,27,28,29,30,31. Las gotas grandes hacen referencia a aquellas con un diámetro mayor a 100 μm, y tienden a sedimentarse rápidamente debido a la gravedad. Por el contrario, las gotas más pequeñas permanecen suspendidas durante períodos de tiempo más prolongados y pueden evaporarse en aerosoles o núcleos de gotas, lo que presenta un riesgo de transmisión a largo plazo. La propagación de virus a través de aerosoles y núcleos de gotitas se denomina «transmisión aérea».
El rango de dispersión de las gotitas para la tos sigue siendo controvertido. Según el trabajo fundamental de Wells, las gotas de 100 μm se depositan a una distancia horizontal de 2 m de quien estornuda29. Sin embargo, Li y colaboradores14 han observado que gotas de 100 μm podrían recorrer distancias de hasta 6,6 m con una velocidad del viento de 2 m/s, pero que esta distancia se veía incrementada en condiciones de sequedad, y Xie y colaboradores30 han encontrado que las gotas podrían viajar más allá de los 6 m basándose en una velocidad de chorro característica para un estornudo de 50 m/s. Incluso con una velocidad de tos más lenta de 10 m/s, las gotas pueden viajar sustancialmente más allá de 2 m. Un trabajo reciente de Bourouiba16 mostró que las actividades espiratorias, como los estornudos y la tos, liberan una turbulenta nube de gas flotante con gotitas suspendidas de varios tamaños. Estas nubes de gas pueden suspender gotitas en el aire hasta distancias de 7 a 8 m antes de perder su impulso. Tanto las trayectorias de las gotas como las tasas de evaporación se ven fuertemente afectadas por la nube de gas. En comparación con las gotas grandes, las gotas más pequeñas son suspendidas por la nube de gas flotante y transportadas a largas distancias. Estas gotitas pueden ser vehículos para patógenos y, por lo tanto, presentan riesgos potenciales para susceptibles huéspedes a ciertas distancias. Un estudio reciente informó que el SARS-CoV-2 puede permanecer viable en aerosoles por un tiempo de hasta 3 horas31. Por lo tanto, comprender el comportamiento en el aire de las gotitas grandes y pequeñas es fundamental para reducir los riesgos de infección y romper la cadena de transmisión de la infección por SARS-CoV-214.
Las trayectorias de las gotas se ven fuertemente afectadas por la aerodinámica. Para comprender mejor la transmisión del SARS-CoV-2, es de vital importancia comprender completamente la dinámica del flujo de aire y de las gotas, incluidas sus interacciones y la evaporación de las gotas. Por ejemplo, en condiciones de alta temperatura y baja humedad relativa (HR), una gota podría evaporarse y encogerse, lo que, a su vez, afecta a su trayectoria y a su destino final14.
El teniente coronel René Pita es jefe del Departamento de Defensa Química de la Escuela Militar de Defensa NBQ.
El teniente coronel (reserva) Juan Domingo es especialista en Defensa NBQ y editor de la página web cbrn.es.
Referencias
- «Emulsions, Foams, Suspensions, and Aerosols: Microscience and Applications», Laurier L. Schramm, Wiley-VCH, 2nd Edition, 2014
- «Fórmula de Stokes», http://www.sc.ehu.es/sbweb/fisica/dinamica/stokes/stokes.html
- «Problemas de física, volumen II, mecánica de fluidos y acústica», E. Gullón de Senespleda y M. López Rodríguez, Librería internacional de Romo, S.L., 3ªEd., 1978
- «Reducing Spray Drift», Vern Hofman & Elton Solseng, AE1210, Reviewed June 2017, https://www.ag.ndsu.edu/publications/crops/reducing-spray-drift/ae1210.pdf
- «Consideraciones sobre el comportamiento de gotas de aspersión», F. R. Leiva, Agronomía Colombiana, volumen 7:110-117, 1990.
- «A Guide to Spray Technology for Dust Control», Bulletin B652A, Spraying Systems Co., https://www.uk.spray.com/literature_pdfs/B652A_Spray_Technology_Dust_Control.pdf
- «Understanding Drop Size», Bulletin B459C, Spraying Systems Co., https://www.spray.com/-/media/dam/industrial/usa/sales-material/product-market-bulletin/b459c_understanding_drop_size.pdf
- «Depósito pulmonar de partículas inhaladas», Ana Fernández Tena y Pere Casan Clarà, Arch Bronconeumol. 2012; 48(7):240–246
- «Inhalation Aerosols: Physical and Biological Basis for Therapy», Anthony J. Hickey, CRC Press, 2ªEd.
- «Embriología del desarrollo de los bronquios y el parénquima pulmonar», María José Acuña Navas y otros, Medicina Legal de Costa Rica, vol. 27 (1), marzo 2010
- «Section V-The Respiratory System», Medical physiology: a cellular and molecular approach / [edited by] Walter F. Boron, Emile L. Boulpaep. , 2nd ed.
- «Información científica-técnica, Enfermedad por coronavirus, COVID-19», Centro de Coordinación de Alertas y Emergencias Sanitarias, Ministerio de Sanidad, Actualización, 15 de enero 2021, https://www.google.es/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwjvh4mD49LuAhVAaRUIHcz5C78QFjAAegQIBRAC&url=https%3A%2F%2Fwww.mscbs.gob.es%2Fprofesionales%2FsaludPublica%2Fccayes%2FalertasActual%2FnCov%2Fdocumentos%2FITCoronavirus.pdf&usg=AOvVaw0e60dQ6xnUJ66KzZGa64xD
- «¿Cómo se propaga la COVID 19 entre las personas?», «Preguntas y respuestas sobre la transmisión de la COVID-19», OMS, actualizado al 20 de octubre de 2020, https://www.who.int/es/news-room/q-a-detail/coronavirus-disease-covid-19-how-is-it-transmitted
- «Dispersion of evaporating cough droplets in tropical outdoor environment», H. Li, F. Yew Leong, G. Xu, Z. Ge, C. Wei Kang & K. Hui Lim, Phys. Fluids 32, 113301 (2020) enviado por Rene
- «The coronavirus pandemic and aerosols: Does COVID-19 transmit via expiratory particles?», S. Asadi, N. Bouvier, A. S. Wexler, & W. D. Ristenpart, Aerosol Sci. 54, 635–638 (2020), https://www.tandfonline.com/doi/pdf/10.1080/02786826.2020.1749229?needAccess=true
- «Turbulent gas clouds and respiratory pathogen emissions: Potential implications for reducing transmission of COVID-19», Lydia Bourouiba, JAMA 323, 1837–1838 (2020).
- «Recognition of aerosol transmission of infectious agents: A commentary», R. Tellier, Y. Li, B. J. Cowling, & J. W. Tang, BMC Infect. Dis. 19, 101 (2019), https://www.researchgate.net/journal/BMC-Infectious-Diseases-1471-2334/publication/330770553_Recognition_of_aerosol_transmission_of_infectious_agents_A_commentary/links/5fc25192299bf104cf88f2ff/Recognition-of-aerosol-transmission-of-infectious-agents-A-commentary.pdf
- «The flow physics of COVID-19», R. Mittal, R. Ni, & J.-H. Seo, J. Fluid Mech. 894, F2-1–F2-14 (2020), https://www.cambridge.org/core/services/aop-cambridge-core/content/view/476E32549012B3620D2452F30F2567F1/S0022112020003304a_hi.pdf/the-flow-physics-of-covid-19.pdf
- «Evidence for probable aerosol transmission of SARS-CoV-2 in a poorly ventilated restaurant», Y. G. Li, H. Qian, J. Hang, X. G. Chen, L. Hong, P. Liang, J. S. Li, S. L. Xiao, J. J. Wei, L. Liu, & M. Kang, (published online 2020). https://www.medrxiv.org/content/10.1101/2020.04.16.20067728v1.full.pdf
- «Airborne transmission of SARS-CoV-2: The world should face the reality», L. Morawska & J. Cao, Environ. Int. 139, 105730 (2020), https://reader.elsevier.com/reader/sd/pii/S016041202031254X?token=20E5A31306FA1888B132065E67E41575AF014E583B2F26435801A286DD486C4D8BE4B4513EF8231A12353B90E22FD501
- «COVID-19 may transmit through aerosol», Juan Wang & Guoqiang Du, Irish Journal of Medical Science, 2020.
- «Avoiding COVID-19:Aerosol guidelines», Matthew J. Evans, Aerosol guidelines, June 8, 2020.
- «Bioaerosol Size Effect in COVID-19 Transmission», Marcelo I. Guzman, Preprints (www.preprints.org), 7 April 2020.
- «The size and the duration of air-carriage of respiratory droplets and droplet-nuclei», J. P. Duguid, Epidemiol. Infect. 44, 471–479 (1946), https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2234804/pdf/jhyg00188-0053.pdf
- «Airborne Contagion and Air Hygiene: An Ecological Study of Droplet Infections», W. F. Wells, Harvard University Press, 1955.
- «Exhaled droplets due to talking and coughing», X. Xie, Y. Li, H. Sun, & L. Liu, J. R. Soc., Interface 6, S703–S714 (2009), https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2843952/pdf/rsif20090388.pdf
- «Violent expiratory events: On coughing and sneezing», L. Bourouiba, E. Dehandschoewercker, & J. W. M. Bush, J. Fluid Mech. 745, 537–563 (2014), https://www.cambridge.org/core/services/aop-cambridge-core/content/view/475FCFCBD32C7DB6C1E49476DB7A7446/S0022112014000883a.pdf/violent-expiratory-events-on-coughing-and-sneezing.pdf
- «Small droplet aerosols in poorly ventilated spaces and SARS-CoV-2 transmission», G. A. Somsen, C. van Rijn, S. Kooij, R. A. Bem, &d. Bonn, Lancet Respir. Med. 8, 658–659 (2020), https://www.thelancet.com/action/showPdf?pii=S2213-2600%2820%2930245-9
- «On air-borne infections: Study II. Droplets and droplet nuclei», W. F. Wells, Am. J. Epidemiol. 20, 611–618 (1934).
- «How far droplets can move in indoor environments—Revisiting the wells evaporation–falling curve», X. Xie, Y. Li, A. T. Y. Chwang, P. L. Ho, & W. H. Seto, Indoor Air 17, 211–225 (2007).
- «Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1», N. van Doremalen, T. Bushmaker, D. H. Morris, M. G. Holbrook, A. Gamble, B. N. Williamson, A. Tamin, J. L. Harcourt, N. J. Thornburg, S. I. Gerber, J. O. Lloyd-Smith, E. de Wit, & V. J. Munster, N. Engl. J. Med. 382, 1564–1567 (2020).