A principios de este mes de enero aparecía una noticia relativa a la tabla periódica de los elementos. En Japón, un equipo de científicos había iniciado la búsqueda del elemento 119, bautizado temporalmente como ununennio (uno uno nueve, en latín), que inauguraría una nueva fila o período en la tabla periódica propuesta en 1869 por el químico ruso Dmitri Ivánovich Mendeléyev. De conseguirse, la primera columna denominada de los metales alcalinos pasaría a tener un nuevo elemento y quedaría así: hidrógeno, litio, sodio, potasio, rubidio, cesio, francio y ununennio1.
Tan sólo hace algo más de un año, el 28 de noviembre de 2016, la IUPAC aprobaba los nombres y los símbolos de los cuatro elementos que completaban la última fila: nihonium (113Nh), moscovium (115Mc), tennessine (117Ts), y oganesson (118Og) 2.
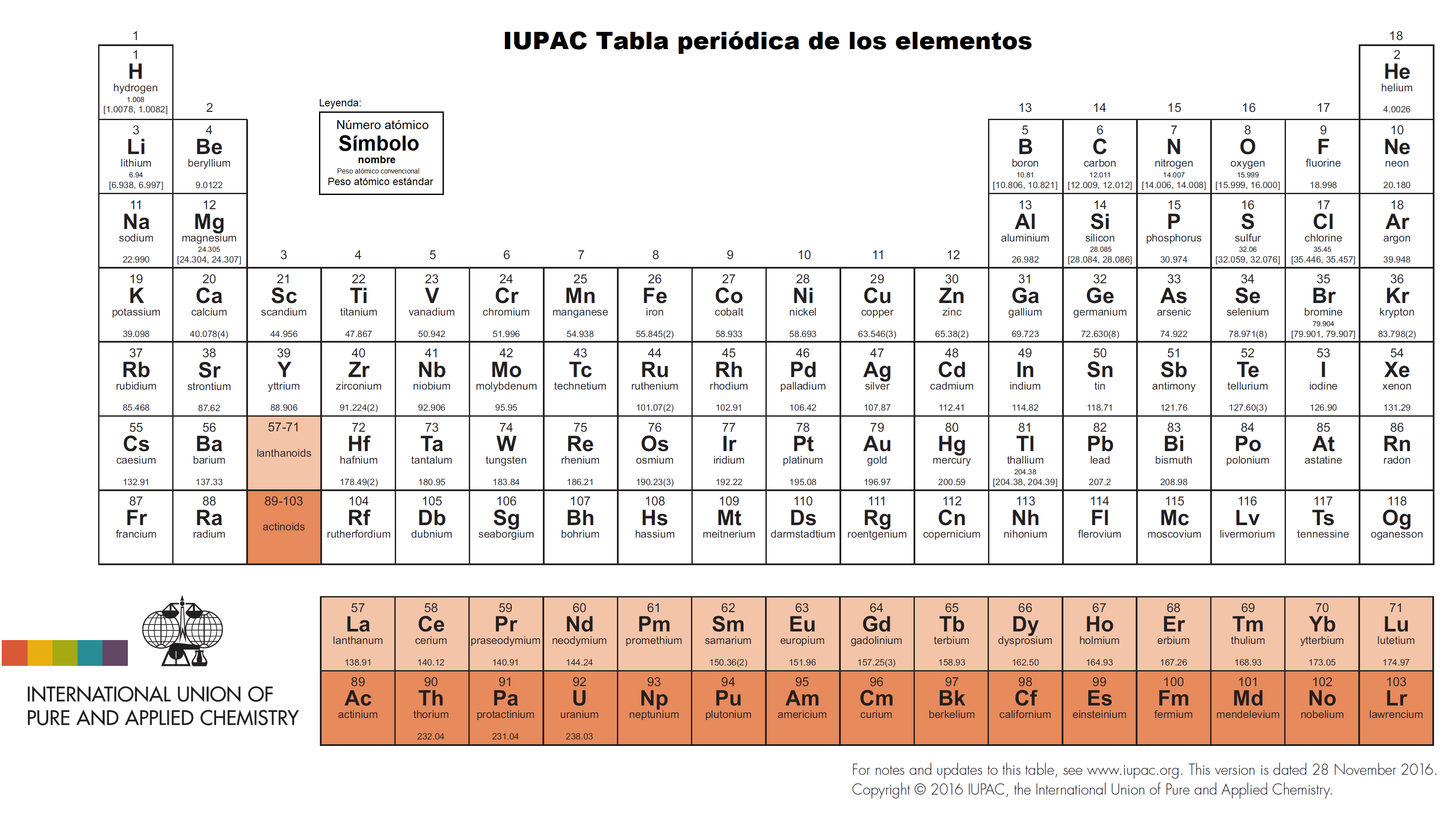
Tabla periódica actual publicada por la IUPAC, https://iupac.org/what-we-do/periodic-table-of-elements/
El uranio, el último elemento
Recordemos que hace ahora un siglo, allá por el año 1918, el uranio era el elemento de mayor número atómico conocido, y la tabla periódica de aquel tiempo presentaba aún siete huecos. La propuesta inicial de Mendeléyev había sufrido con el paso del tiempo algunas importantes variaciones debido al descubrimiento de nuevos elementos gracias al espectroscopio (especialmente el eka-aluminio o galio, el eka-silicio o escandio, y el eka-boro o germanio, que corroboraban las hipótesis de Mendeléyev), al descubrimiento de los gases nobles (una nueva hilera), la inclusión en un solo hueco de las tierras raras del lantano, al descubrimiento de la radiactividad (y de las series radiactivas que acababan en el plomo),y al reconocimiento de los isótopos y del número atómico (que permitió a Henry Moseley enunciar su ley y ordenar los elementos en función de su número atómico).
Hace aproximadamente un siglo la tabla periódica de Mendeléyev tenía un aspecto parecido al que se muestra en la siguiente figura:
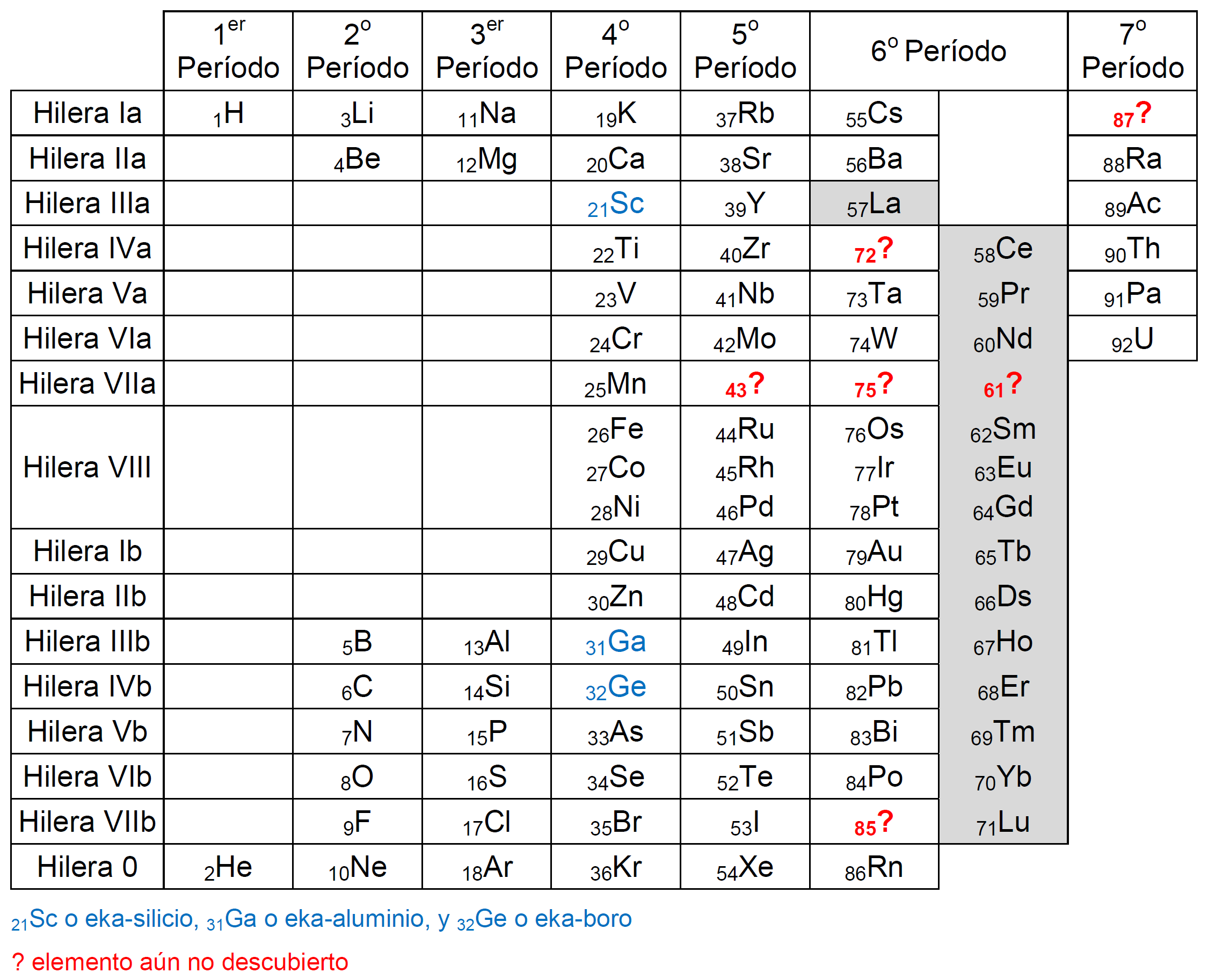
Tabla periódica con los elementos descubiertos hasta 1918 cuando se descubrió el protactinio
El último elemento añadido a esta tabla había sido el elemento 91, denominado protactinio (Pa), situado justo antes del elemento 92, el uranio (U), considerado el último elemento, descubierto muchos años antes, en 1789, por Martin Heinrich Klaproth.
En 1913 Kasimir Fajans y Oswald Helmuth Göhring durante sus estudios sobre la cadena de decaimiento del 92U-238 descubrieron un nuevo elemento radiactivo con un periodo de semi-desintegración muy corto, al que denominaron «brevium» precisamente por su «breve» periodo de semi-desintegración (este elemento resultaría ser 91Pa-234m). En 1918 Otto Hahn y Lise Meitner descubrieron un elemento radiactivo, el 91Pa-231, procedente también del decaimiento del uranio que fue finalmente identificado como el elemento de número atómico 91 y recibió el nombre de protactinio (también se atribuye el descubrimiento del protactinio, en 1918, a Frederich Soddy y John A. Cranston). El aislamiento del protactinio no sería posible hasta el año 1934, cuando el químico nuclear alemán Aristid von Grosse consiguió preparar óxido de protactinio, Pa2O5, que convirtió en ioduro de protactinio, PaI5 y posterior transformación de éste en protactinio metálico (2 PaI5 → 2 Pa + 5 I2, proceso Van Arkel-De Boer). El protactinio se comportaba tal y como había descrito Mendeléyev, quien le había dado el nombre provisional de eka-tantalio. Se conocen 30 isótopos de protactinio, todos ellos radiactivos, con periodos de semidesintegración que van desde 53 nanosegundos a 3,276 × 104 años.
La tabla presentaba aún seis huecos (43?, 61?, 72?, 75?, 85? y 87?) que serían rellenados con nuevos elementos antes del descubrimiento del elemento 93, que abriría la búsqueda de nuevos elementos sintéticos (y radiactivos), más allá del uranio.
Los seis huecos
- El elemento 72, denominado hafnio (Hf), tiene una controvertida historia asociada a su descubrimiento.Varios investigadores, incluído el químico francés Georges Urbain, afirmaron haber descubierto el elemento de manera independiente pero más tarde se comprobó que se habían confundido3. En 1923 Georg Karl von Hevesy y Dirk Coster, con el asesoramiento del físico danés Niels Henrik David Bohr, utilizaron la espectroscopía de rayos x para estudiar la disposición electrónica de la corteza externa de circonio y su análisis les permitió identificar el hafnio4.
El hafnio tiene 44 isótopos conocidos, cinco de ellos estables (72Hf-176 = 5,26%, 72Hf-177 = 18.60%, 72Hf-178 = 27,28%, 72Hf-179 = 13.62% y 72Hf-180 = 35.08%.). Uno de los isótopos radiactivos, el 72Hf-174 = 0,16%, tiene un período de semi-desintegración tan grande, 2,0 × 1015 años, que se contabiliza su abundancia en la corteza terrestre junto a la de los isótopos estables. A pesar de que el hafnio no es un elemento escaso o raro, no fue descubierto hasta 1923 debido a su estrecha asociación con circonio. Varios científicos sospecharon de la presencia junto al circonio de un nuevo elemento pero nadie fue capaz de separarlos e identificarlo dado que el mineral de circonio contenía cerca de 50 veces más circonio que hafnio. El nombre de hafnio proviene de Hafnia, el nombre en latín de la ciudad de Copenhague (Dinamarca), en honor a Niels Bohr que había nacido en Copenhague, y al hecho de que los trabajos se habían llevado a cabo en el «Niels Bohr Institute» de Copenhague4.
- El elemento 75, denominado renio, fue descubierto en Berlín en 1925 por el equipo del matrimonio formado por Walter Noddack e Ida Tacke Noddack, que estaban buscando el eka-manganeso, elemento de número atómico 43 (que sería el tecnecio) y el dvi-manganeso, elemento de número atómico 75 (que sería el renio)3,5. En 1925, publicaron el documento (Zwei neue Elemente der Mangangruppe, Chemischer Teil), alegando que habían descubierto ambos, y llamaron masurio y renio, respectivamente a estos nuevos elementos. Fue confirmado el descubrimiento del renio, pero no el del masurio, y luego de descubierto el elemento 43, el nombre de masurio no fue aceptado por cuestiones nacionalistas, pues hacía referencia a Masuria, una región de la antigua Prusia oriental.
Calcularon y predijeron algunas de las propiedades químicas y físicas del dvi-manganeso, elemento de numero atomico 75, y en 1925, mediante el empleo de varias técnicas analíticas, consiguieron concentrar del orden de 100 000 veces un mineral de gadolinio en una pequeña muestra que les permitió estudiar e identificar espectroscópicamente el elemento de número atómico 75, al que bautizaron con el nombre de renio, en honor al río Rin (en alemán, Rhein)4,5.
Se conocen 45 isótopos del renio, y sólo uno de ellos es estable, el 75Re-185, que aporta el 37,40 % a la cantidad total de renio que se encuentran en la tierra. El 75Re-187, radiactivo, con un período de semi-desintegración muy largo, 4,35 × 1010 años, aporta el 62.60 % restante. Los demás isótopos son radiactivos, tienen periodos de semi-desintegración muy cortos, y se obtienen artificialmente4.
- El honor del descubrimiento del elemento 43, el eka-manganeso de Mendeléyev fué para los italianos Carlo Perrier y Emilio Gino Segrè, que lo obtuvieron 12 años más tarde, en 1937, en la Universidad de California en Berkeley. Llamaron al nuevo elemento Tecnecio (Tc) para reflejar el hecho de que había sido sintetizado artificialmente como subproducto de una reacción nuclear3,5.
Muchos científicos afirmaron haber descubierto el elemento de número atómico 43 e incluso le asignaron nombres como davyum, illmenium, lucium y nipponium, pero sus descubrimientos resultaron ser erróneos. Por esa época Enrico Fermi había transformado un elemento químico en otro por bombardeo con deuterones, 1H-2, núcleos de hidrógeno que tienen 1 protón y 1 neutrón. Esta transmutación artificial permitía transformar un elemento en otro, era lo que habían estado buscando durante mucho tiempo los antiguos alquimistas que intentaron sin éxito transformar el plomo en oro4,5.
El tecnecio fue descubierto mediante espectroscopía de rayos X por Walter Noddack e Ida Tacke, en Berlín, en un mineral de platino enviado desde Colombia, pero sus resultados no pudieron confirmarse. En 1937, Emilio Gino Segrè y Carlo Perrier que conocían el trabajo de Fermi, pensaron que si el elemento 43 no se podía encontrar, quizás podría fabricarse utilizando la técnica de Fermi. Con ayuda de un ciclotrón bombardearon molibdeno (42Mo) con deuterones, de modo que añadieron un protón al núcleo de molibdeno, y así sintetizarón el elemento 43 al que denominaron tecnecio (Tc), que en griego significa artificial4,5.
Se conocen 47 isótopos del tecnecio, ninguno de ellos estable, todos radiactivos, y la mayoría de ellos producidos artificialmente en ciclotrones (aceleradores de partículas) y reactores nucleares. Los isótopos del tecnecio cubren un amplio rango de masa, desde el 43Tc-85 al 43Tc-118, la mayoría con un periodo de semi-desintegración muy corto. Para establecer el peso atómico del tecnecio se utilizaron los dos isótopos con periodo de semidesintegración más largo 43Tc-98 (4,2 × 106 años) y 43Tc-99 (2,111 × 105 años)4.
- El elemento 87 fue descubierto en 1939, en París (Francia), por Marguerite Perey, asistente de Marie Curie, que le dió el nombre de Francio (Fr), en honor a su país3. Perey descubrió la secuencia de desintegración radiactiva del radio en actinio y luego en otros isótopos desconocidos, uno de los cuales identificó como 87Francio-2234:
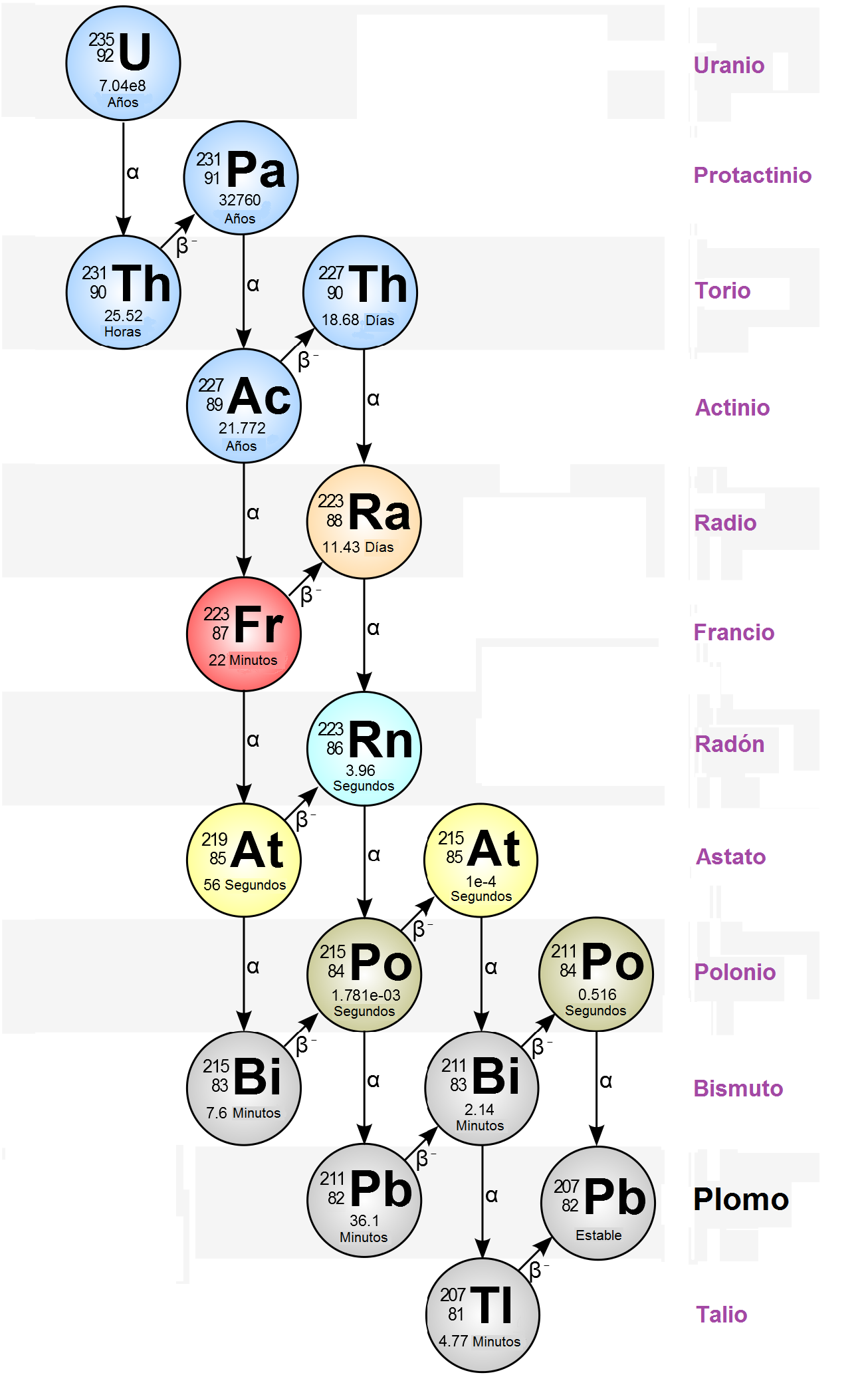
Los 33 isótopos del francio (desde el 87Fr-201 al 87Fr-232) son radiactivos, habiéndose utilizado el 87Fr-223, que tiene el periodo de semi-desintegración más largo (del orden de 20 minutos) para la determinación de su peso atómico. El 87Fr-223 es el único isótopo del francio que se puede encontrar de manera natural, como producto de la desintegración de elementos inestables4.
- El elemento 85, el astato, fue descubierto en 1940 por Emilio Gino Segrè3. Los 41 isótopos conocidos del astato son radiactivos, con vidas medias que van desde 125 nanosegundos hasta 8,1 horas. El isótopo 85As-210, el isótopo más duradero con un periodo de semidesintegración de 8,1 horas, se utilizó para determinar el peso atómico del astato. El 85As-210 decae por decaimiento alfa en 83Bi-206 o por captura de un electrón en 84Po-210. A principios de la segunda guerra mundial, en 1940, Dale Raymond Corson, K. R. Mackenzie y Emilio Gino Segrè crearon un elemento nuevo con 85 protones con ayuda de un ciclotrón. Aunque la guerra interrumpió su trabajo, reanudado éste, en 1945, consiguieron el astato al bombardear con partículas alfa (núcleos de helio) de alta energía un blanco de 83Bi-209. Este el método que todavía hoy se utiliza para la producción de pequeñas cantidades de 85As-211 (más dos neutrones)4.
- Finalmente el elemento 61, que recibió el nombre de Prometio (Pm), fue descubierto por tres químicos del «Oak Ridge National Laboratory», Jacob A. Marinsky, Lawrence E. Glendenin, y Charles D. Coryell, entre los productos de la fisión nuclear.3 Su descubrimiento no se produjo de una forma deliberada, sino como un resultado más del descubrimiento de la fisión nuclear.
El prometio tiene 64 isótopos con periodos de semi-desintegración que van desde dos milisegundos a más de 17 años, y todos ellos se producen artificialmente a partir de los residuos remanentes en los reactores nucleares. Aunque el prometio fue descubierto en 1944, sus autores no reclamaron el descubrimiento hasta 1946. Parece ser que fue la esposa de Coryell, Grace Mary Coryell, quien sugirió el nombre de Prometio por ser Prometeo, según la mitología griega, quien robó el fuego de los dioses para dárselo a los hombres.4
El elemento 93, descendiente del uranio
Tras los descubrimientos del neutrón en 1932 por James Chadwick y de la radiactividad artificial a principios de 1934 por Irene and Frederic Joliot-Curie, Fermi y su equipo inicaron en 1934 el bombardeo sistemático de todos los elementos químicos a su alcance con el objetivo de producir nuevas especies radiactivas y nuevas reacciones nucleares6,8,9.
En aquel entonces los neutrones se producían mediante la reacción nuclear entre una partícula α, 2He-4, (procedente de 88Ra o de otro emisor α) y berilio. El berilio, elemento monoisotópico, 4Be-9, se mezclaba bien con el emisor α, y en la reacción se emitían neutrones de diferentes energías (diferentes velocidades). La captura del neutrón es más probable con neutrones de baja velocidad, de modo que suele utilizarse parafina como moderador, para conseguir neutrones de baja velocidad (proceso conocido como termalización)7:
![]()
Cuando Fermi y su grupo (E.Fermi, E.Amaldi, O.D’Agostino, F.Rasetti y E. G. Segrè) llegaron a uranio, encontraron varias actividades nuevas, todas ellas con emisiones beta (observaron al menos cinco emisores de radiación b, que diferían en sus periodos de semi-desintegración de 10 segundos, 40 segundos, 13 minutos, 90 minutos y alrededor de un día. Determinaron que la radiación no era debida al uranio, ni a elemento alguno por debajo del uranio, así que Fermi propuso que el núcleo de uranio había capturado un neutrón y luego había iniciado una secuencia de decaimiento de tipo beta, con la producción de nuevos elementos, el elemento 93 e incluso el elemento 94, los primeros elementos artificiales. Este estraño descubrimiento atrajo la atención de todos, incluso de la prensa popular6,7,8,9.
El mayor problema, para Fermi y sus posibles nuevos elementos artificiales eran los supuestos físicos y químicos, que luego se desmostrarían falsos, que contradecían los resultados observados5,6.
Los físicos siempre habían observado que los núcleos, incluso los núcleos radioactivos eran bastante estables, de modo que cuando se producía un decaimiento radiactivo u otra reacción nuclear, los cambios siempre eran pequeños. Los resultados obtenidos por Fermi con los neutrones eran consistentes con ello, pues había encontrado que con los elementos más ligeros, el neutrón podía golpear el núcleo y sacar de él un protón, «reacción (n, p)», o incluso, una partícula α,»reacción (n,α)», y que con los elementos más pesados, la reacción el neutrón era siempre una «captura radiativa», «reacción (n,γ)». Si un nuevo núcleo artificial era radiactivo, siempre se producía un decaimiento con emisión de una partícula beta para formar el siguiente elemento con número atómico superior5,6.
| |
|
| |
No es de extrañar pues, que Fermi, cuando encontró en el uranio varias nuevas actividades todas ellas con emisiones beta, pensase que se debían a elementos con número atómico superior al del uranio6. Sobre la base de la tabla periódica de aquel entonces pensó que el primer elemento transuránico, con número atómico 93, debía ser químicamente como el renio (y lo denominó eka-renio, Eka-Re), el elemento 94 debería ser como el osmio (y lo denominó eka-osmio, Eka-Os) y así sucesivamente10.
Sin embargo, los químicos habían supuesto incorrectamente que los elementos transuránicos (situados más allá del uranio) tendrían que tener la misma química que los elementos de transición. Habían considerado al uranio y a los elementos que le precedían como elementos de transición, puesto que, químicamente, se asemejaban poco entre sí, y se asemejaban más a los elementos de transición situados por encima de ellos, y estaba totalmente asumido que los elementos transuránicos serían también elementos de transición, que ocuparían sus lugares por debajo de la tercera fila de los elementos de transición (es decir, en el 7º período, debajo del Re, Os, Ir, etc.)5,6.
Puesto que las fuentes de neutrones (generalmente radón mezclado con berilio en polvo) eran débiles, las nuevas actividades de tipo beta no eran mucho más fuertes que la radiactividad natural del uranio y de sus productos de decaimiento. Fermi separó las nuevas actividades del uranio mediante coprecipitación de éstas con compuestos de los metales de transición, lo que apoyaba la idea de que se trataban de elementos transuránicos, pero no pudo obtener una evidencia inequívoca6,10.
En 1935, el equipo de Fermi abandonó estas investigaciones, que fueron retomadas, en Berlín, por el equipo de Otto Hahn, Lise Meitner, y Fritz Strassmann, que mejoró el método de separación de Fermi y comenzó un laborioso proceso para conocer a fondo las actividades en el precipitado. En 1937 habían encontrado una cantidad impresionante de nuevas especies radiactivas que se asignaron a tres procesos distintos, dos procesos que mostraban una amplia secuencia de decaimientos de tipo β (que luego se demostraría eran procesos de fisión), y un tercer proceso, muy diferente, atribuido a la captura resonante de un neutrón lento6:
| 1 | 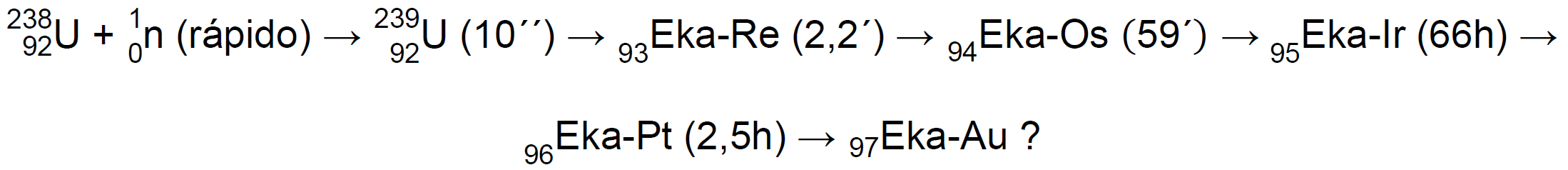 |
| 2 | |
| 3 | |
En 1940, en el laboratorio de física de la Universidad de California, en Berkeley, Edwin Mattison McMillan y un estudiante de postgrado, Philip Hauge Abelson bombardearon óxido de uranio con neutrones de alta velocidad mediante el empleo de un ciclotrón. Su experimento mostró la presencia que un nuevo elemento que exhibía propiedades químicas y físicas similares al uranio, con estados de oxidación de +4 y +6. Bombarderon 92U-238 con neutrones de alta energía, que produjeron 92U-239 que, a su vez, se descompuso en 93Np-239 con emisión de radiación β4,7,11:
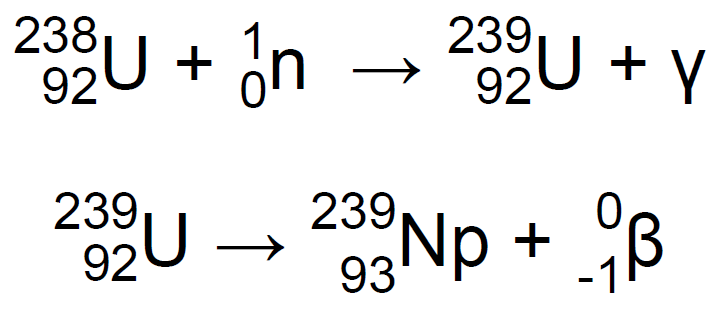
En 1940 pudieron confirmar su descubrimiento y publicar los datos. Puesto que el elemento de número atómico 93 iba a continuación del elemento de número atómico 92, el uranio, McMillan decidió darle el nombre de Neptunio (Np), por ser el planeta Neptuno el siguiente al planeta Urano. El trabajo de McMillan y Abelson se interrumpió durante la segunda guerra mundial y fue luego retomado por Arthur C. Wahl y Joseph W. Kennedy, que determinaron las reacciones físicas que dan lugar a la formación del neptunio4,11.
El neptunio tiene 23 isótopos todos ellos radiactivos con periodos de semi-desintegración que van desde los microsegundos a los 2,144 × 106 años del isótopo 93Np-2374.
Hoy en día, el 93Np-237 se produce en los reactores nucleares mediante las reacciones12:
| En un 70%: | En un 30%: |
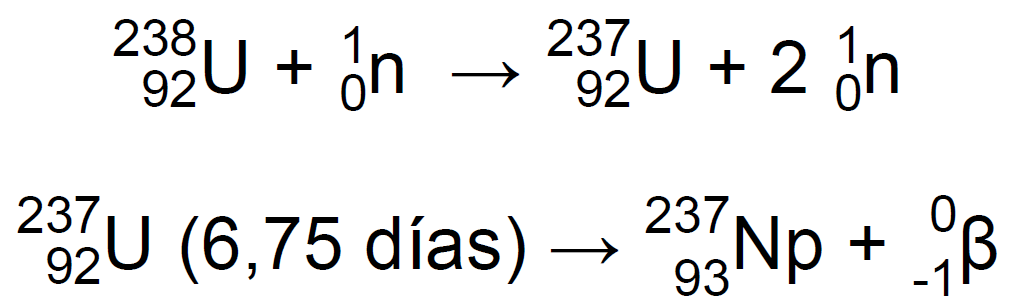 |
 |
Sorprendentemente, las propiedades químicas del neptunio eran similares a las del uranio y no a las del renio, como sugería la tabla periódica de la época. Después de descubrir el neptunio, McMillan empezó a buscar el supuesto eka-osmio producido por el decaimiento del 93Np-239 que obviamente correspondería al nuevo elemento 94. Sin embargo no pudo encontrarlo. Hoy sabemos la causa, el largo periodo de semi-desintegración del 94Pu-239, de 24 000 años, provocaba una radiación tan baja en su muestra, que la detección resultaba imposible7,11. El honor del descubrimiento del plutonio quedaría para G. T. Seaborg, E. M. McMillan, J. W. Kennedy, y A. C. Wahl, que lo conseguirían al bombardear uranio con deuterones en un ciclotrón, y obtener 94Pu-23811,12:
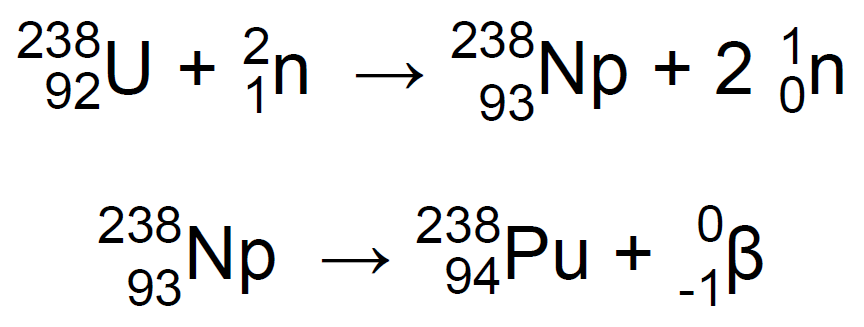
El 94Pu-238 tiene un periodo de semidesintegración de tan solo 88 años12.
Referencias
- «La tabla periódica se asoma a una nueva fila por primera vez en la historia», https://elpais.com/elpais/2018/01/04/ciencia/1515101255_058583.html
- «IUPAC Announces the Names of the Elements 113, 115, 117, and 118», https://iupac.org/iupac-announces-the-names-of-the-elements-113-115-117-and-118/
- «The Periodic Table-Its Story and Its Significance», Eric R. Scerri, Oxford University Press, 2007.
- «The history and use of our earth»s chemical elements-a reference guide», Robert E. Krebs, Greenwood Press, 2006.
- «Chemical Sciences in the 20th Century- Bridging Boundaries», C. Reinhardt, Wiley-VCH, 2001.
- «The Search for Transuranium Elements and the Discovery of Nuclear Fission», Ruth Lewin Sime, Phys. perspect. (2000) 2: 48-62.
- «On Beyond Uranium-Journey to the end of the Periodic Table», Sigurd Hofmann, Taylor & Francis, 2002
- «Artificial Radioactivity Produced by Neutron Bombardment», E. Fermi, E. Amaldi, O. D’Agostino, F. Rasetti, E. G. Segrè, Proc. R. Soc. Lond. A 1934 146 483-50.
- «Artificial radioactivity produced by neutron bombardment—II», E. Amaldi, O. D’Agostino, E. Fermi, B. Pontecorvo, F. Rasetti, E. G. Segrè, Proc. R. Soc. Lond. A 1935 149 522-558.
- «Nuclear Fission and Transuranium Elements-50 Years Ago», Glenn T. Seaborg, Journal of Chemical Education 1989 66 (5), 379.
- «The chemistry of the actinide and transactinide elements», L.R. Morss, N.M. Edelstein & J.Fuger, fourth edition, volumes 1–6, Springer, 2010.
- «Radiochemistry and Nuclear Chemistry», Gregory R. Choppin, Jan-Olov Liljenzin & Jan Rydberg, Elsevier Inc., 2013.