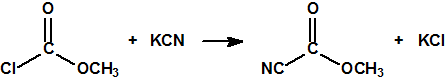El término «trilon» es una marca comercial registrada de la BASF Aktiengesellschaft. La fábrica de la IG Farben (BASF, Badische Anilin- und Soda-Fabrik) en Ludwigshafen utilizó en 1936 el término «trilon» para referirse al ácido nitrilotriacético (NTA, Nitrilo Triacetic Acid) o mejor a la sal sódica de dicho ácido1. Hoy en día, la empresa BASF ofrece bajo la marca trilon® una amplia gama de productos2 con diferentes agentes quelantes, por ejemplo, «trilon A», en base al ácido nitrilo triacético, NTA; «trilon B», en base al ácido etilendiaminotetraacético, EDTA; «trilon C», en base al ácido dietilentriaminopentaacético, DTPA; «trilon D», en base al ácido hidroxietiletilendiaminotriacético, HEDTA, y «trilon M», en base al ácido metilglicinadiacético, MGDA.
Sin embargo el término «trilón» también les resultará familiar a algunos especialistas en Defensa NBQ, en relación con los nombres recibidos por el tabún (trilon-83)3,5,7,9,10,11,12,13,14,15,16,17 y el sarín (trilon-46)5,8,9,11,12,13,15,16. Incluso parece que existirían más trilones, como el somán (trilon-300)8,9,11,13,16, el etil-tabún (trilon-32)7, y etil-sarin (trilon-113)13. No todos los autores coinciden en los códigos utilizados e incluso se pueden encontrar contradicciones entre algunos de los nombres codificados.
¿Por qué esta denominación de trilones para algunos agentes neurotóxicos?. Pues parece ser que los dirigentes del Reich alemán, quisieron estudiar y fabricar estos agentes químicos sin llamar la atención de los servicios de inteligencia aliados. Como la fábrica I.G. de Ludwigshafen fabricaba un producto para el tratamiento de las aguas denominado «trilon» o «trilone», decidieron codificar estas sustancias denominándolas también como «trilones» con el fin de que pasaran desapercibidas.
El tabún, «trilon-83»7,18
En 1932, en la Universidad de Berlín, el químico alemán Willy Lange (1900-1976) y su asistente Gerda von Krueger (1907-d1970) habían sintetizado varios miembros de la familia de los fosforofluoridatos de dialquilo, por ejemplo, los fosforofluoridatos de dimetilo, de dietilo, de propilo y de dibutilo, y habían observado que sus efectos tóxicos eran similares a los producidos por la nicotina, utilizada también como insecticida, pues afectaba la transmisión nerviosa por su parecido con la acetilcolina.
En los laboratorios de la I.G. Farbenindustrie, en Leverkusen, Gerard Schrader (1903-1990) y su equipo procedieron también a la síntesis de más de 2000 compuestos con fósforo y flúor buscando en ellos propiedades insecticidas, y encontraron que los fluoruros y difluoruros dialquilamidofosfóricos resultaban útiles como tales. En 1935, Gerhard Schrader y Otto Bayer (1902-1982) presentaban una patente (US2146356) sobre estos nuevos insecticidas (Dialkylaminophosphorous fluorides and a process for preparing the same):
 |
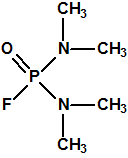 |
| fluoruros dialquilamidofosfóricos (X corresponde a oxígeno o azufre, y R corresponde a un radical alquílico con 1-3 átomos de carbono) | Dimefox, fluoruro de N, N, N’, N’-tetrametil diamido fosforilo, CAS 115-26-4 |
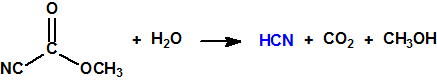
En la búsqueda de estructuras similares que aportasen mejores resultados, Schader decidió incorporar el grupo el grupo ciano (-CN), en vista de que el ácido cianhídrico era tóxico. En diciembre de 1936 trató el dicloruro dimetilamidofosfórico con cianuro sódico en etanol en un intento de obtener el correspondiente dicianuro dimetilamidofosfórico:
Sin embargo, el compuesto que obtuvo fue el N,N-dimetil fosforamidocianidato de O-etilo, al que denominó Le-100 («Le» provenía de Leverkusen), y que más tarde pasaría a denominarse tabún:
El tabún resultó altamente tóxico para los animales de laboratorio y por tanto de poco valor como insecticida. Su enlace con las colinesterasas resultaba extremadamente fuerte y en estas circunstancias no podían realizar la ruptura de la acetilcolina, que se acumulaba afectaba la transmisión nerviosa.
Un decreto nazi de 1935 requería que se informase de todo descubrimiento de interés militar, y así lo hizo la IG Farben. A instancias de Leopold von Sicherer y Wolfgang Wirth de la División 9 del Departamento de Ensayo y Desarrollo de Armas se solicitaba un informe detallado de la sustancia Le-100. Schrader describió la síntesis y propiedades del Le-100 a Leopold von Sicherer, al coronel Ernst Rüdiger von Brüning, de la División 9, y a Hermann Van der Linde, jefe del Laboratorio de Protección de Gas del Ejército. Estos quedaron impresionados por las propiedades del Le-100, que se convertía en el primer agente neurotóxico de guerra con el nombre de tabún, palabra inventada sin significado alguno. No obstante para ocultar la identidad del tabún se emplearon otros nombres en clave, como Gelan, trilon 83 o T-83 o Stoff 100, y más tarde recibiría por los aliados el acrónimo de GA.
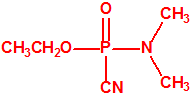 |
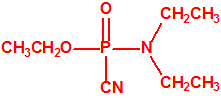 |
| tabún, «trilon-83» | etil tabún, «trilon-32» |
El sarín, «trilon-86»7
A finales de 1938, en Elberfeld, Schrader desarrolló una nueva familia de insecticidas reemplazando el grupo ciano y el grupo amida del tabún, con un átomo de flúor y un radical alquílico unido directamente al átomo de fósforo. Uno de estos compuestos, el metilfosfonofluoridato de O-isopropilo resultó ser sorprendentemente tóxico para los insectos. Las pruebas de toxicidad posteriores que Eberhard Gross realizó sobre mamíferos demostraron que la nueva sustancia era mucho más tóxica que el tabún y no podía utilizarse como insecticida comercial. Schrader asignó a esta sustancia el código 146, y, a comienzos de 1939, Gross envió su informe toxicológico a la Oficina de Guerra Alemana. Schrader tuvo que describir la síntesis y propiedades de la sustancia 146 al personal del Laboratorio de Protección de Gas del Ejército, que inmediatamente asignó un nutrido grupo de químicos para su estudio, y desarrollo de un proceso de fabricación industrial.
Los oficiales del Laboratorio de Protección de Gas del Ejército denominaron a la sustancia 146 como SARIN, acrónimo derivado de las letras de los nombres de las cuatro personas claves implicadas en su desarrollo: los químicos Gerhard Schrader y Otto Ambros de la IG Farben y el coronel Ernst Rüdiger von Brüning y Hermann Van der Linde de la Oficina de Artillería del Ejército. Al igual que ocurrió con el tabún, al sarín se le asignarían diversos nombres en clave para ocultar su identidad, como trilon 46, T144 o Gelan III, y más tarde recibiría por los aliados el acrónimo de GB.
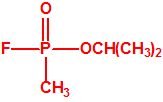 |
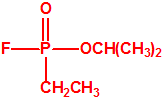 |
| sarín, «trilon-46» | etil-sarín, «trilon-113» |
El somán, «trilon-300»4,6
A principios de 1943, el coronel Oskar Schmidt del Army Ordnance Office pidió a Richard Kuhn (1900-1967), director del Instituto de Química en el Kaiser Wilhelm Institute for Medical Research, en Heidelberg, analizar los efectos de los agentes nerviosos sobre los sistemas nerviosos periférico y central. Kuhn, químico alemán, había recibido el premio Nobel de Química de 1938 por su trabajo en carotenoides y vitaminas.
Cuando Kuhn y su equipo comenzaron las investigaciones en 1943 sobre el mecanismo de acción de los agentes nerviosos, no solo conocían los trabajos de Gerard Schader sobre el tabún y el sarín, también conocían los trabajos del fisiólogo inglés Henry Hallett Dale (1875-1968) y del fisiólogo alemán Otto Loewi (1873-1961) acerca del papel clave que desempeña la acetilcolina en la parte parasimpática del sistema nervioso autónomo y del sistema nervioso periférico.
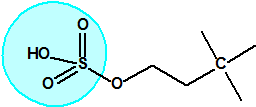
El equipo de Kuhn estudió la relación entre la estructura química molecular y los efectos biológicos sobre la colinesterasa. Los experimentos realizados en ratas indicaban que sustancias químicas con una cierta similitud estructural a la acetilcolina causaban de manera similar una inhibición de la colinesterasa. La síntesis de una serie de ésteres del ácido sulfúrico con similitud estructural a la acetilcolina no condujo a buenos resultados así que se decidió continuar los ensayos con ésteres del ácido metilfluorofosfórico:
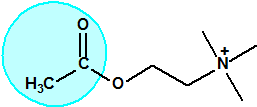 |
 |
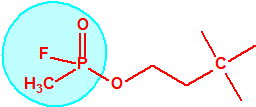 |
| acetilcolina | sulfato de alquilo | metilfluorofosfonato de alquilo |
Un colaborador de Kuhn, el químico alemán Konrad Henkel (1915-1999) realizó la síntesis de unos diez compuestos por esterificación del metilfosfonildifluoruro con diversos alcoholes. La reacción con el 3,3-dimetil-1-butanol (CAS 624-95-3) produjo un inhibidor de la colinesterasa que resultó más tóxico que el tabún, pero como no existían reservas del 3,3-dimetil-1-butanol, y resultaba demasiado difícil de producir, se optó por el 3,3-dimetil-2-butanol (CAS 464-07-3), también conocido como alcohol pinacolílico, que produjo el metilfosfonofluoridato de O-pinacolilo, más conocido como somán.
 |
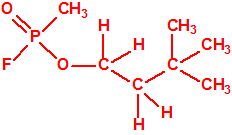 |
| somán, «trilon-300» | isómero del somán, más tóxico que éste |
Los isómeros del somán
Cuando Henkel realizo la esterificación del metilfosfonildifluoruro con diversos alcoholes buscaba una cierta similitud estructural con la acetilcolina. El 3,3-dimetil-1-butanol se asemeja mucho estructuralmente a la acetilcolina, pero al no estar fácilmente disponible se optó por el 3,3-dimetil-2-butanol, ambos de fórmula empírica C6H14O. Existen 17 alcoholes alifáticos de fórmula empírica C6H14O, y además algunos de ellos tienen un átomo de carbono asimétrico, de modo que presentan isómeros ópticos. La siguiente tabla resume las estructuras, nombres y números CAS de estos alcoholes, así como las estructuras, nombres y números CAS de sus correspondientes metilfosfonofluoridatos:
| alcohol C6H14O pm: 102,17 | CAS | trilon C7H16FO2P pm: 182,17 | CAS | |
| 1 | 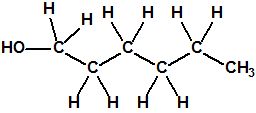 1-hexanol 1-hexanol |
111-27-3 |  |
113548-89-3
MS/IR/RMN |
| 2 | 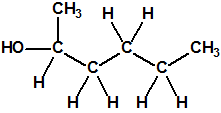 2-hexanol, butil metil carbinol * 2-hexanol, butil metil carbinol * |
626-93-7 | 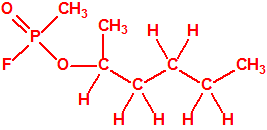 |
13172-12-8
MS/IR |
| 3 | 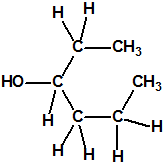 3-hexanol, etil propil carbinol * 3-hexanol, etil propil carbinol * |
623-37-0 | 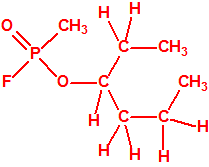 |
959218-68-9
MS/IR |
| 4 | 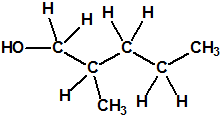 2-metil-1-pentanol * 2-metil-1-pentanol * |
105-30-6 | 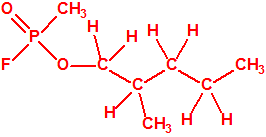 |
333416-05-0
MS/IR |
| 5 | 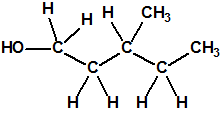 3-metil-1-pentanol * 3-metil-1-pentanol * |
589-35-5 | 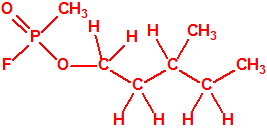 |
199850-60-7
MS/IR |
| 6 | 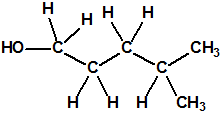 4-metil-1-pentanol 4-metil-1-pentanol |
626-89-1 | 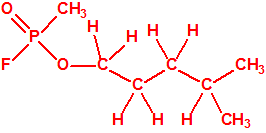 |
959010-20-9
MS/IR |
| 7 | 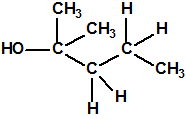 2-metil-2-pentanol, dimetil propil carbinol 2-metil-2-pentanol, dimetil propil carbinol |
590-36-3 | 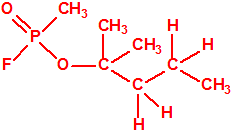 |
959088-49-4
|
| 8 | 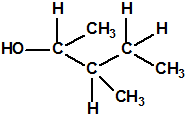 3-metil-2-pentanol, sec-butil metil carbinol * 3-metil-2-pentanol, sec-butil metil carbinol * |
565-60-6 | 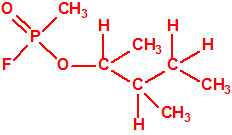 |
1005239-89-3
MS |
| 9 | 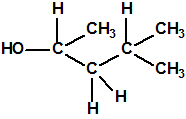 4-metil-2-pentanol, 3-MIC, iIsobutil metil carbinol, MAOH, MIBC * 4-metil-2-pentanol, 3-MIC, iIsobutil metil carbinol, MAOH, MIBC * |
108-11-2 | 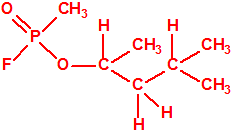 |
352-53-4
MS/IR |
| 10 | 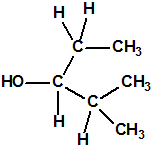 2-metil-3-pentanol, etil isopropil carbinol * 2-metil-3-pentanol, etil isopropil carbinol * |
565-67-3 | 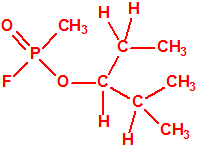 |
345260-66-4
MS/IR |
| 11 | 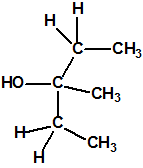 3-metil-3-pentanol, dietil metil carbinol 3-metil-3-pentanol, dietil metil carbinol |
77-74-7 | 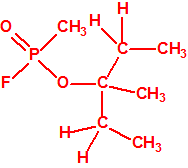 |
959083-58-0
|
| 12 | 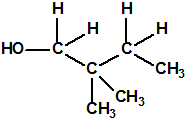 2,2-dimetil-1-butanol 2,2-dimetil-1-butanol |
1185-33-7 | 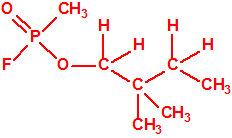 Metilfosfonofluoridato de O-(2,2-dimetil-butilo) Metilfosfonofluoridato de O-(2,2-dimetil-butilo) |
SIN CAS |
| 13 | 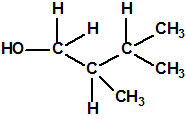 2,3-dimetil-1-butanol * 2,3-dimetil-1-butanol * |
19550-30-2 | 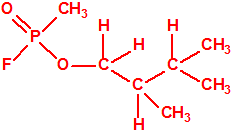 |
83563-66-0
|
| 14 | 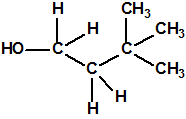 3,3-dimetil-1-butanol más toxico 3,3-dimetil-1-butanol más toxico |
624-95-3 | 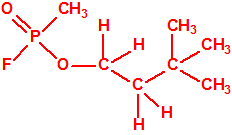 |
660-21-9
MS/IR/RMN |
| 15 |  2,3-dimetil-2-butanol 2,3-dimetil-2-butanol |
594-60-5 | 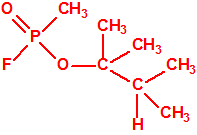 |
959026-90-5
|
| 16 | 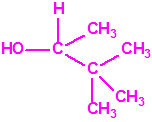 3,3-dimetil-2-butanol, tert-buti metil carbinol, alcohol pinacolílico * 3,3-dimetil-2-butanol, tert-buti metil carbinol, alcohol pinacolílico * |
464-07-3 | 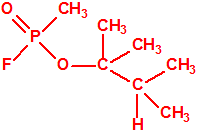 |
96-64-0
MS/IR/RMN |
| 17 | 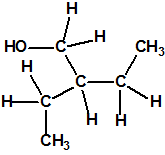 2-etil-1-butanol, isohexilalcohol, 2-etilbutil alcohol 2-etil-1-butanol, isohexilalcohol, 2-etilbutil alcohol |
97-95-0 |  |
126204-48-6
MS/IR |
* indica la presencia de isómeros ópticos
Todos los alcoholes tienen asignado número CAS, algunos incluso para sus isómeros ópticos, pero parece que el metilfosfonofluoridato de O-(2,2-dimetil-butilo), ni tiene asignado número CAS, ni registrado espectro de masas, espectro infrarrojo o espectro de resonancia magnética nuclear (según la «OPCW Central Analytical Database«, enero 2017). Recordemos que todos estos metilfosfonofluoridatos tienen el mismo peso molecular que el somán, propiedades fisicoquímicas y toxicológicas parecidas, y podrían ser algunos de esos «trilones» que no conocemos.
El metilfosfonofluoridato de O-(3,3-dimetilbutilo), que resultó ser más tóxico que el somán, metilfosfonofluoridato de O-(1,2,2-trimetilpropilo), también podría ser uno de esos «trilones» no conocidos.
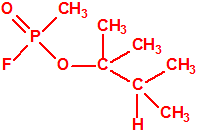 |
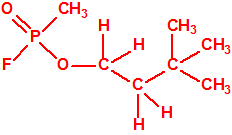 |
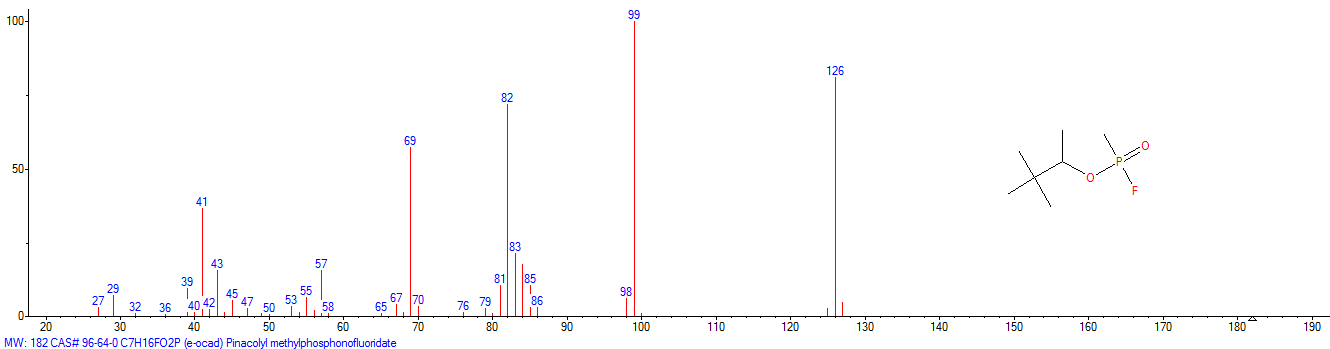 |
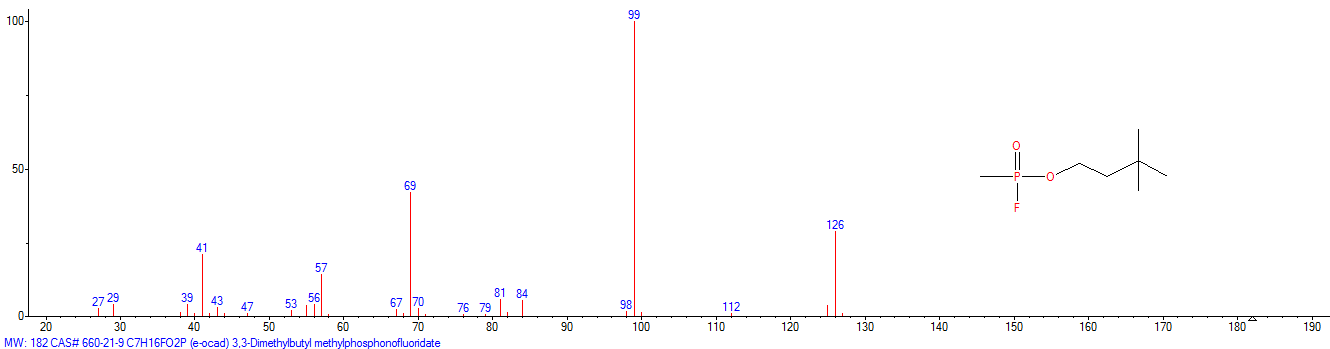 |
| «trilon-300», somán, metilfosfonofluoridato de O-(1,2,2-trimetilpropilo), CAS 96-64-0 | metilfosfonofluoridato de O-(3,3-dimetilbutilo), CAS 660-21-9 |
En realidad, si considerásemos «trilones» a los agentes nerviosos o neurotóxicos similares al tabún, al sarín o al somán, tendríamos que hablar de las familias de los alquil fosfonofluoridatos de O-alquilo y de los N,N´-dialquil fosforamidocianidatos de O-alquilo, es decir, de las listas 1A.1 y 1A.2 de la Convención para la Prohibición de las Armas Químicas, que suponen más de 20000 y más de 50000 sustancias químicas, respectivamente.
Referencias
- «Complexing Agents-A Study of Short-term Toxicity, Catalytic Oxidative Degradation and Concentrations in Industrial Waste Waters», Kari Pirkanniemi, http://epublications.uef.fi/pub/urn_isbn_978-951-27-0782-9/urn_isbn_978-951-27-0782-9.pdf
- http://worldaccount.basf.com/wa/NAFTA~en_US/Catalog/ChemicalsNAFTA/pi/BASF/Brand/trilon/brand_top/
- «Cholinesterases and anticholinesterase agents», G. B. Koelle, O. Eichler& A. Farah, Springer-Verlag , 1963
- «Fluorine Chemistry at the Millennium-Fascinated by fluorine», R.E. Banks, Elsevier, 2000
- «A History of Chemical Warfare», Kim Coleman, Palgrave MacMillan, 2005
- «Neurosciences and Research on Chemical Weapons of Mass Destruction in Nazi Germany», Florian Schmaltz, Journal of the History of the Neurosciences, 15:186–209, 2006
- «War of nerves: chemical warfare from World War I to Al-Qaeda», Jonathan B. Tucker, Pantheon Books, 2006
- «A Laboratory History of chemical Warfare Agents», Jared B. Ledgard, 2006
- «Elservier´s Dictionary of Chemoetymology», Alexander Senning, Elsevier, 2007
- «Counter-terrorism for emergency responders», Robert Burke, CRC Press, Taylor & Francis Group, 2007
- «Medical Aspects of Chemical Warfare», Shirley D. Tuorinsky, Medical Department of the Army, 2008
- «Chemical Warfare Agents-Chemistry, Pharmacology, Toxicology, and Therapeutics», James A.Romano&others, CRC Press, 2008
- «Handbook of Chemical and Biological Warfare Agents», D. Hank Ellison, CRC Press, 2Ed., 2008
- «The A to Z of Nuclear, Biological, and Chemical Warfare», B.C. Garrett & J. Hart, The Scarecrow Press, 2009
- «The Kaiser Wilhelm Society under National Socialism», S. Heim, C. Sachse & M. Walker, Cambridge University Press, 2009
- «Eintrag zu trilone». In: Römpp Online. Georg Thieme Verlag, abgerufen am 1. April 2014.
- «Secret Science-a century of poison warfare and human experiments», Ulf Schmidt, Oxford University Press, 2015
- «Drug Discovery-A History», Walter Sneader, John Wiley & Sons, 2005