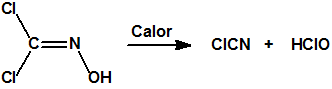
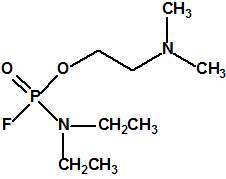
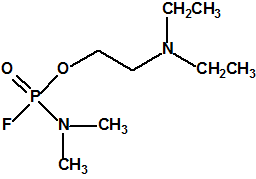
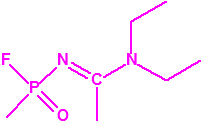
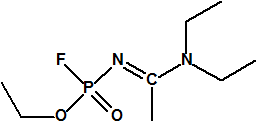
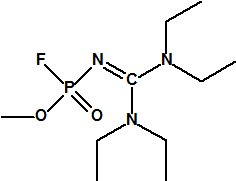
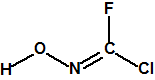
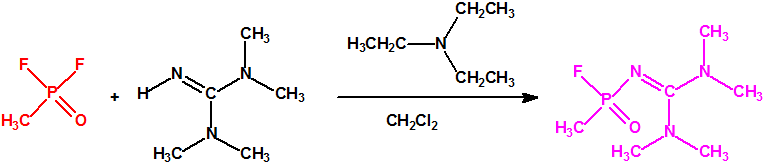
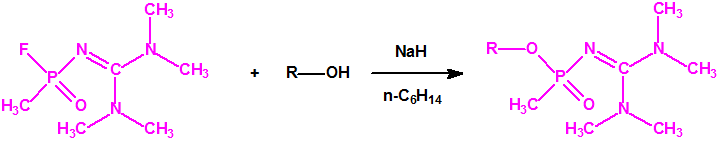
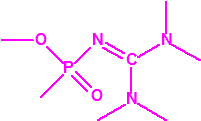
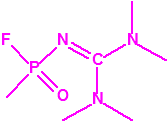
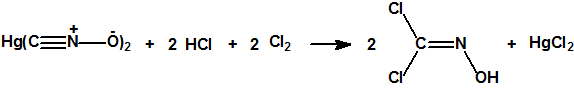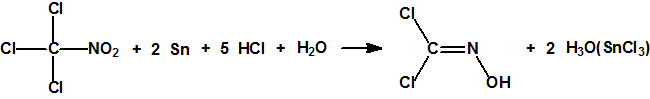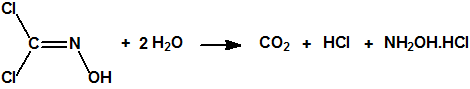A las cinco de la tarde del día 22 de abril de 1915, en Ypres, las tropas alemanas liberaban del orden de 168 toneladas de cloro contenidas en unas 5730 bombonas metálicas (unas 1600 cargadas con 40 kg de cloro cada una, y las otras 4130 cargadas con 20 kg de cloro). El cloro liberado, formó una inmensa nube amarillo verdosa y el viento arrastró estos vapores, más densos que el aire, hacia las trincheras donde se encontraban las fuerzas argelinas y francesas. Las tropas, sorprendidas y sin medios de protección, trataban de escapar corriendo hacia su retaguardia, en la misma dirección que los vapores de cloro, aumentado con ello su exposición a los mismos. Las tropas alemanas que no esperaban semejante efecto, no estaban preparadas para la explotación del éxito y desaprovecharon el factor sorpresa, que ya no se repetiría en posteriores ocasiones, pues las tropas aliadas estarían preparadas con pañuelos mojados con agua u orina, con los que se tapaban la nariz y la boca1.
A la vista de estos hechos parece claro que las armas químicas son especialmente eficaces empleadas por sorpresa, en grandes cantidades (para conseguir una elevada concentración), contra tropas sin protección (que no se encuentren dispersas).
Para conseguir el efecto tóxico deseado, ya sea incapacitante o letal, es necesario que las víctimas inhalen o reciban la dosis apropiada. En el caso de inhalación de una sustancia química tóxica la dosis letal en función del tiempo, por ejemplo, LCt50 es la dosis letal resultado de la inhalación de una determinada concentración durante un determinado tiempo, que produciría la muerte al 50 por ciento de la población expuesta. Cuanto más pequeño sea el valor de la LCt50 menor concentración o menor tiempo de exposición se requiere para conseguir los mismos efectos letales.
Por ejemplo, si la LCt50 para el sarín fuese 100 mg×min/m3, en un ambiente con una concentración de sarín de 1000 mg/m3, bastarían 6 segundos de inhalación para alcanzar el valor de la dosis letal 50 en función del tiempo. En cambio, para el cloro, con una la LCt50 de 10 000 mg×min/m3, sería necesario un tiempo de inhalación de 10 minutos (600 segundos). Aplicando el mismo razonamiento, la inhalación durante un minuto en un entorno contaminado, requiere una concentración de sarín de tan solo 100 mg/m3, para alcanzar la dosis letal 50 en función del tiempo, mientras que se requiere una concentración de 10 000 mg/m3 de cloro para alcanzar la dosis letal 50 correspondiente.
En campo abierto, las condiciones meteorológicas influyen mucho en el movimiento y dispersión de la nube tóxica, de ahí que se requieran grandes cantidades del agente químico de guerra, y que el objetivo no esté disperso, para conseguir una concentración suficientemente alta en la zona del objetivo, que inhalada el tiempo conveniente permita se alcance la dosis letal.
Guerra química y agentes químicos de guerra
La guerra química se define como el empleo de agentes químicos para matar, herir, o incapacitar durante un periodo de tiempo significativo, hombres y animales, y prohibir o dificultar el uso de áreas, instalaciones o material, o defenderse contra este empleo2.
También se define agente químico como una sustancia química que se pretende usar en operaciones militares para matar, herir seriamente, o incapacitar, por medio de sus efectos fisiológicos. El término excluye los agentes antidisturbios cuando se emplean el mantenimiento del orden, los herbicidas, los fumígenos y los incendiarios2.
La Convención sobre las Armas Químicas (CAQ) en su artículo II, «Definiciones y criterios», entiende por «armas químicas», conjunta o separadamente3:
- Las sustancias químicas tóxicas o sus precursores, salvo cuando se destinen a fines no prohibidos por la presente Convención, siempre que los tipos y cantidades de que se trate sean compatibles con esos fines;
- Las municiones o dispositivos destinados de modo expreso a causar la muerte o lesiones mediante las propiedades tóxicas de las sustancias especificadas en el apartado a) que libere el empleo de esas municiones o dispositivos; o
- Cualquier equipo destinado de modo expreso a ser utilizado directamente en relación con el empleo de las municiones o dispositivos especificados en el apartado anterior.
Y entiende por «sustancia química tóxica»: «Toda sustancia química que, por su acción química sobre los procesos vitales, pueda causar la muerte, la incapacidad temporal o lesiones permanentes a seres humanos o animales. Quedan incluidas todas las sustancias químicas de esa clase, cualquiera que sea su origen o método de producción, y ya sea que se produzcan en instalaciones, como municiones o de otro modo»3. A los efectos de la aplicación de la CAQ, las sustancias químicas tóxicas respecto de las que se ha previsto la aplicación de medidas de verificación están enumeradas en Listas incluidas en un Anexo B sobre sustancias químicas.
En caso de una liberación intencionada las sustancias químicas tóxicas penetrarían en el organismo básicamente por dos vías:
- Por vía inhalatoria, en forma de vapor, gas o aerosol, la sustancia química tóxica ejercería su acción a través del sistema respiratorio con efectos rápidos y peligrosos.
- Por vía cutánea, en forma líquida, gaseosa o aerosol, la sustancia química tóxica ejercería su acción a través de la piel, heridas y ojos.
Aunque cada sustancia química, en función de sus propiedades, ejerce su acción tóxica preferentemente por una de estas dos vías, dependiendo fundamentalmente de las condiciones meteorológicas existentes, podrían hacerlo por ambas vías.
Las sustancias químicas de bajo peso molecular y/o bajo punto de ebullición tienen una volatilidad elevada, y una baja persistencia, y son consideradas «agentes no-persistentes», que actúan fundamentalmente por vía inhalatoria, durante un periodo de tiempo relativamente breve. Por el contrario, las sustancias químicas de alto peso molecular y/o alto punto de ebullición tienen una volatilidad reducida, y una alta persistencia, son consideradas «agentes persistentes», que actúan fundamentalmente por vía cutánea, durante un periodo de tiempo bastante prolongado, contaminando personal, medios y terreno.
Con el empleo de agentes químicos de guerra, se busca, además de matar, herir, o incapacitar al enemigo, obligar a éste a emplear medios de protección, disminuyendo con ello sus capacidades operativas. Para su empleo en operaciones militares los agentes químicos se clasifican en:
- Agentes químicos no persistentes, que actúan fundamentalmente por inhalación durante un breve período de tiempo, que tienen como objetivo causar bajas y abrir una brecha en las posiciones enemigas, de modo que transcurrido un cierto tiempo, esa zona pueda ser utilizada por las tropas propias sin necesidad de utilizar equipo de protección, y
- Agentes químicos persistentes, que actúan fundamentalmente por contacto, cuyo objetivo es impedir o limitar la utilización del material y/o el terreno al contaminar durante un largo período de tiempo los mismos.
Durante la Primera Guerra Mundial los alemanes contemplaban en su doctrina el empleo de proyectiles de iperita y de proyectiles «rompe-máscaras», seguidos de proyectiles de fosgeno, en una táctica desarrollada por el teniente coronel Georg Bruchmüller, conocida como «cruces multicolores» (Buntkreuz), o «disparos multicolores» (Buntshiessen). La doctrina de empleo de armas químicas de los británicos era algo distinta, pues consistía en realizar ataque químicos sobre unidades seleccionadas, con vistas a debilitarlas y desmoralizarlas, a través de un hostigamiento continuado, que producía un efecto devastador sobre la moral de las tropas1,4,5.
En los años treinta, el Servicio de Guerra Química de EE. UU. incluía en su doctrina el empleo de aeronaves que volasen a baja altura y a baja velocidad, para el rociado con iperita que permitiese contaminar rápidamente grandes extensiones velocidad. Se comprobó la necesidad de conseguir gotas de un cierto tamaño (no muy pequeño) para que el viento no las arrastrase y disminuir además su evaporación. La solución fue «espesar» la iperita (y otros agentes), con algún espesante, como por ejemplo, poliestireno y metacrilato de metilo, para aumentar el tamaño y viscosidad de las gotas1,6.
Los japoneses desarrollaron una doctrina de empleo de agentes persistentes, iperita y lewisita, que consistía en lanzarlos por detrás de las líneas de las tropas enemigas cuando éstas iniciaban su retirada, con el fin de ralentizarla1,7.
En 1984, durante la guerra Irán-Iraq, los iraquíes siguiendo su doctrina de empleo de armas químicas, contaminaron con iperita las rutas de suministro de las unidades a vanguardia, cortando así su apoyo logístico. Más tarde ante la ofensiva iraní recurrieron al empleo de tabún, un agente no-persistente, para abrir brechas y recuperar objetivos1,8,9.
Atendiendo a sus efectos fisiológicos los agentes químicos de guerra se pueden clasificar en:
- Agentes sofocantes o neumotóxicos
- Agentes tóxicos sanguíneos o cianogénicos
- Agentes vesicantes o dermotóxicos
- Agentes neurotóxicos o nerviosos, que se subdividen en agentes de la serie G (básicamente, no-persistentes) y agentes de la serie V (persistentes)
- Agentes incapacitantes
Todos los agentes químicos de guerra, sean del tipo que sean, presentan además efectos psicológicos muy importantes. El miedo y el horror que inspiran alteran la moral y el estado anímico de las personas (personal militar y civil) provocando incluso pánico.
El empleo de armas químicas está considerado hoy como una flagrante violación de la legalidad internacional y un crimen contra la humanidad, de modo que a nadie en su sano juicio, ni siquiera en la situación más adversa, se le ocurriría emplear armas químicas, y menos contra personal civil, especialmente niños.
La CAQ
Después del empleo de armas químicas durante la Primera Guerra Mundial, y ante la opinión pública favorable a la prohibición de las armas químicas, el 17 de junio de 1925, treinta y ocho naciones firmaron el Protocolo de Ginebra de 1925, denominado «Protocolo relativo a la prohibición del empleo en la guerra de gases asfixiantes, tóxicos o similares y de medios bacteriológicos», que prohibía «el empleo en la guerra de gases asfixiantes, tóxicos o similares. Algunos países que ratificaron el Protocolo lo hicieron con la reserva de que la prohibición desaparecería en el momento en que el enemigo o sus aliados no respetasen el Protocolo. Además el Protocolo prohibía el uso de armas químicas y armas biológicas, pero no decía nada acerca de su producción, su almacenamiento o su transferencia1.
Tras varios años de negociaciones, en la Conferencia de Desarme, en Ginebra, finalizó la redacción del texto de la Convención sobre las Armas Químicas (su título completo es Convención sobre la prohibición del desarrollo, la producción, el almacenamiento y el empleo de armas químicas y sobre su destrucción), que se abrió a la firma el 13 de enero de 1993, en París, y entró en vigor el 29 de abril de 1997, 180 días después de haber sido depositado el 65º instrumento de ratificación (Hungría).
Con el fin de asegurarse de que se toman las medidas necesarias para el cumplimiento de esos ambiciosos objetivos, la CAQ prevé un complejo régimen de verificación. Con sus actividades de inspección in situ y de seguimiento de los datos, el sistema permite verificar que las actividades realizadas en los Estados Partes son coherentes con los objetivos de la CAQ y con el contenido de las declaraciones presentadas a la Organización para la Prohibición de las Armas Químicas (OPAQ). Las inspecciones son cruciales para la aplicación de la CAQ, pudiéndose distinguir tres tipos de inspección: las inspecciones ordinarias de las instalaciones relacionadas con las armas químicas y de las instalaciones de industria química, que emplean ciertas sustancias químicas «de doble uso» (es decir, que pueden ser empleadas para fines tanto pacíficos como prohibidos); las inspecciones por denuncia, notificadas con muy poca antelación, que pueden ser efectuadas en cualquier lugar de cualquier Estado Parte que revista preocupación en relación con el no cumplimiento para otro Estado Parte; y las investigaciones sobre el presunto empleo de armas químicas. Todo lo referente a las inspecciones está detallado en el anexo sobre la aplicación y la verificación (anexo de verificación) que muchas veces parece ignorarse3,10.
Las armas químicas en Siria
Recordemos que el 14 de septiembre de 2013 el Secretario General de la ONU comunicaba haber recibido de Siria, conforme estipula el artículo XXIII de la CAQ, su solicitud de adhesión a la Convención de Armas Químicas (CAQ) y que también ese día, EE.UU. y Rusia hacían público un acuerdo para destruir el arsenal químico sirio y evitar así una acción de castigo solicitada insistentemente tras los incidentes de Ghouta, el 21 de agosto de 2013. En este acuerdo, EE.UU. y Rusia se comprometían a preparar y remitir al Consejo Ejecutivo de la OPAQ un borrador con “procedimientos especiales” para la destrucción rápida del programa sirio de armas químicas y su rigurosa verificación. Este acuerdo incluía la destrucción de toda la capacidad química siria antes de la primera mitad del año 2014, es decir, antes del 30 de junio de 201411.
El 14 de octubre de 2013 la Republica Árabe Siria pasó a ser el Estado Parte número 190 en la Convención para la prohibición de las Armas Químicas (CAQ). En consecuencia, no más tarde de transcurridos treinta días, el 24 de octubre de 2013, presentaba formalmente a la OPAQ su declaración inicial, de carácter confidencial, acerca de su programa de armas químicas, y también un plan para la destrucción de las mismas, en el que indicaba que la única forma de destruir su arsenal químico de manera rápida y segura conforme a las condiciones recogidas por la CAQ era realizando la misma fuera de su territorio11.
El 15 de noviembre de 2013 el Consejo Ejecutivo de la OPAQ aprobaba el plan detallado de destrucción para eliminar el arsenal sirio de armas químicas de la «manera más rápida y segura», que tenía como objetivo más importante completar la destrucción antes de la primera mitad de 2014, según lo que había establecido en la decisión del Consejo Ejecutivo de la OPAQ y en la resolución del Consejo de Seguridad de la ONU 2118 (2013), ambas de 27 de septiembre de 201311.
Las primeras noticias sobre el arsenal químico sirio hablaban de unas 1300 toneladas de iperita, sarín y VX, sin detallar más, con un texto ambiguo que daba a entender que las 1300 toneladas se referían a agentes químicos de guerra (sustancias de lista 1A de la CAQ).
Hoy sabemos que el arsenal declarado de sustancias químicas se reducía a 20,25 toneladas de iperita, 540 toneladas de metilfosfonildifluoruro (DF), precursor de Lista 1, 290 toneladas de sustancias de Lista 2, 110 toneladas de sustancias de Lista 3, 398 toneladas de sustancias no incluidas en las Listas de la OPAQ, algunas ni siquiera incluidas en el Grupo Australia, y una cantidad no detallada de alcohol isopropílico, que aunque está incluido en lista alguna forma parte del sistema binario del sarín. No declaró poseer ni sarín, ni VX11.
Después de algo más de dos años, el lunes 4 de enero de 2016, se anunciaba que había finalizado la destrucción de todas las sustancias químicas declaradas por la República Árabe Siria, retiradas de su territorio en 2014. A pesar de ello sus problemas con las armas químicas están aún lejos de concluir11.
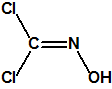
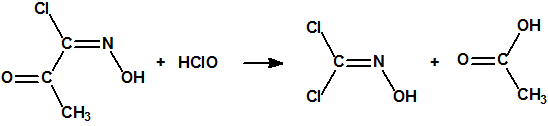
Para el gobierno sirio, las armas químicas, lejos de ser una solución a sus problemas, han resultado ser uno de sus principales quebraderos de cabeza. Desde que se inició el conflicto sirio en 2011, se han realizado por diferentes entidades y países, de uno y otro bando, numerosas denuncias acerca del empleo de armas químicas, sarín y cloro fundamentalmente, y alguna vez iperita12,13.
Puesto que Siria no había ratificado aún la CAQ, las primeras investigaciones sobre algunas de las múltiples denuncias sobre el empleo de armas químicas se llevaron a cabo mediante el Mecanismo del Secretario General (MSG) para la investigación del supuesto empleo de armas químicas y biológicas, puesto en marcha por el Secretario General de la ONU, Ban Ki-moon, el 21 de marzo de 2013, tras la denuncia del Gobierno sirio acerca del empleo de armas químicas en la localidad de Khan Al Asal. A la investigación sobre el incidente de Kahn Al Asal ocurrido el 19 de marzo de 2013, se acabaron incorporando otros incidentes: el de Sheik Maqsood, ocurrido el 13 de abril, el de Saraqeb, ocurrido el 29 de abril, el de Ghouta, ocurrido el 21 de agosto 2013, el de Bahhariyeh, ocurrido el 22 de agosto, el de Jobar, ocurrido el 24 de agosto, y el de Ashrafiah Sahnaya, ocurrido el 25 de agosto14.
El 13 de diciembre se presentaba el informe A/68/663–S/2013/735 que confirmaba el empleo armas químicas (sarín), no solo en la zona de Ghouta (Damasco) el 21 de agosto de 2013 como se concluyó en el documento A/67/997-S/2013/553, sino también en menor escala en Jobar, el 24 de agosto de 2013, Saraqueb, el 29 de abril de 2013, Ashrafiah Sahnaya, el 25 de agosto de 2013 y Khan al-Asal, el 19 de marzo de 2013. El informe no aportaba información sobre quién era el responsable de los hechos15,16.
El 29 de abril de 2014, el Director General de la Organización para la Prohibición de Armas Químicas (OPAQ) anunció la creación de una Misión para la Determinación de los Hechos en relación con el supuesto empleo de armas químicas en Siria (Fact-Finding Mission)17.
La OPAQ dio a conocer el primer informe sobre la misión para la determinación de los hechos en relación con el supuesto empleo de cloro en la República Árabe Siria, el 16 de junio de 2014 (S/1191/2014) y el 10 de septiembre de 2014, dio a conocer el segundo informe (S/1212/2014), que concluía que los testimonios aportados por 37 testigos constituían una «confirmación convincente» (compelling confirmation), de que se había empleado, sistemática y repetidamente, una sustancia química tóxica como método de guerra, y que, con un «alto grado de confianza» (high degree of confidence), esa sustancia química tóxica era cloro. El informe NO indicaba quién había podido ser el autor de los hechos. El tercer informe, fechado el 18 de diciembre de 2014 (S/1230/2014) no decía nada nuevo que no dijeran los anteriores informes. Simplemente proporcionaba una descripción más detallada sobre la labor realizada y el proceso que condujo a los resultados presentados en su segundo informe. El documento concluía de nuevo que, con un «alto grado de confianza», se había empleado cloro como método de guerra, y recalcaba que su trabajo, consistente con su mandato, no incluía la cuestión de la atribución de responsabilidad por la presunta utilización18.
Dado que la Misión de Determinación de los Hechos de la OPAQ no tenía el mandato de llegar a una conclusión sobre la atribución de responsabilidad por el empleo de armas químicas, el consejo de seguridad de Naciones Unidas, aprobaba en su 7501ª sesión, celebrada el 7 de agosto de 2015, Resolución 2235 (2015), la creación del Mecanismo Conjunto de Investigación de la OPAQ y las Naciones Unidas (JIM, Joint Investigative Mechanism) para identificar en la mayor medida posible a las personas, entidades, grupos o gobiernos que hayan empleado sustancias químicas como arma, incluido el cloro o cualquier otra sustancia química tóxica, en la República Árabe Siria o que hayan organizado o patrocinado su empleo o participado en él de cualquier otro modo19. El 17 de noviembre de 2016 el Consejo de Seguridad en Resolución 2319 (2016) renovaba el mandato del JIM por otro año, pero el 24 de octubre de 2017, primero, y luego el 17 de noviembre, rechazaba las propuestas para prorrogar su mandato por otro año más.
Durante su mandato el Mecanismo de Investigación Conjunto de la Organización para la Prohibición de Armas Químicas (OPAQ) y de la ONU (JIM) presentó siete informes, y concluyó que, en cuatro ocasiones, desde 2015 a 2107, el Gobierno sirio era responsable de tres ataques con cloro y uno con sarín. Esto indicaría que o bien en 2013 el Gobierno sirio no habría declarado la totalidad de su programa químico o bien lo habría conservado una pequeña capacidad de producción de agentes neurotóxicos (sarín) y habría vuelto a utilizar cloro, una sustancia química industrial tóxica, como arma química.
Terrorismo químico
La CAQ está muy cerca de conseguir la destrucción de la totalidad de las armas químicas declaradas, pues solo le queda la destrucción de dos instalaciones sirias, en vías de destrucción y la finalización de la destrucción de las armas química de Estados Unidos prevista para el año 2023.
Además tan sólo quedan cuatro estados por ratificar la CAQ y conseguir así la membresía total. Estos cuatro estados son Corea del Norte, Egipto, Israel (la ha firmado pero no la ha ratificado) y Sudán del Sur.
A la vista de los acontecimientos más recientes, una de las mayores amenazas para la CAQ es el empleo terrorista de las sustancias químicas tóxicas ya sea para cometer asesinatos más o menos selectivos, o para sembrar el pánico y el terror entre la población civil.
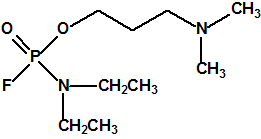
Los supuestos agentes «novichok» son según dicen del orden de 5-7 veces más tóxicos que el VX, es decir, utilizados de manera similar al VX, se requerirían cantidades del orden de 5-7 menores para producir la misma dosis letal. Para facilitar los cálculos supongamos que la dosis letal LD50 para el VX es de 10 µg/Kg (vía dérmica), y supongamos que nuestras personas tienen un peso de 100 Kg; entonces la LD50 sería de 1mg/persona. Supongamos, para facilitar los cálculos, que la densidad del VX fuese 1 mg/mL (la densidad real es 1,008 mg/mL a 20°C), entonces 1 mg de VX sería equivalente a 1 µL de VX, es decir, la LD50 sería de 1 µL/persona (El 50% de las personas de 100 Kg de peso que entrasen en contacto con 1 µL de VX fallecerían). Observe que desde el punto de vista clínico se considera que 20 gotas equivalen a 1 mL, es decir que 1 gota sería del orden de 50 µL, y que la LD50 calculada para el VX es de tan solo 1 µL, algo así como la cabeza de un alfiler.
Si en vez de VX empleásemos un supuesto agente «novichok», la cantidad requerida sería mucho menor de 1 µL, y de emplear esa cantidad la letalidad obtenida sería mucho mayor.
En cuanto a cómo hacer llegar la dosis a nuestros individuos de modo que los daños colaterales fueran mínimos, existen numerosas posibilidades, función sobre todo de su toxicidad y persistencia.
Los últimos casos de asesinatos selectivos con armas químicas, el asesinato, en el aeropuerto de Kuala Lumpur (Malasia), de Kim Jong-un, en 2017, empleando VX y el intento de asesinato del ex espía ruso Sergei Skripal y su hija Yulia, en Salisbury (Reino Unido), en 2018, empleando un agente «novichok», demuestran que, en este tipo de acciones, el empleo de agentes químicos no es más efectivo que el empleo de armas de fuego, pero eso sí, provocan el caos a nivel organizativo y político.
Doctrina Pá Ná
A la vista del empleo de armas químicas durante el conflicto sirio, antes y después de la adhesión de la República Árabe de Siria a la Convención, ya sea con agentes químicos de guerra, sarín e iperita, o con sustancias químicas industriales tóxicas, cloro, parece que la doctrina de guerra química siria, nada tiene que ver con la de los alemanes durante la Primera Guerra Mundial, pareciéndose algo a la doctrina inglesa durante ese mismo conflicto, que se enfocaba sobre todo en el aspecto psicológico sobre los combatientes. Esta doctrina podríamos denominarla «Doctrina Pá Ná», pues los agentes químicos lejos de afectar a los combatientes enemigos, afectan a civiles, y sobre todo a niños, con lo que en vez de conseguir algún tipo de ventaja o beneficio militar lo que consigue es la repulsa e indignación del resto del mundo.
Los hechos corroboran, desde el punto de vista táctico, el supuesto empleo «pá ná» de armas químicas por parte del Gobierno sirio.
Después de múltiples denuncias sobre incidentes químicos, el Gobierno sirio ratifica la Convención a finales del año 2013, y evita una intervención militar internacional de castigo, que era inminente.
La guerra continua y los incidentes químicos siguen produciéndose, a pesar de que a principios de 2016 se diera por finalizada la destrucción de todas las sustancias químicas declaradas en su arsenal químico.
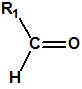
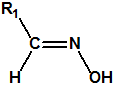
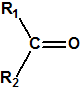
Cuando parece que el curso de la guerra es favorable al gobierno sirio, tiene lugar, el 4 de abril de 2017, el incidente de Khan Shaykhun, un ataque con armas químicas (con sarín o con una sustancia parecida al sarín, según el informe de la Misión de Determinación de los Hechos de la OPAQ), que causó al menos 86 muertos, todos ellos civiles, según el Observatorio Sirio de Derechos Humanos (OSDH)20.
Según Estados Unidos y los grupos armados opositores al gobierno sirio, dos aviones del gobierno bombardearon la ciudad en su totalidad, especialmente los centros de concentración de civiles como clínicas y hospitales. Las Autoridades sirias y Rusia alegaron que se había bombardeado un almacén donde los rebeldes, que controlaban Khan Shaykhun, guardaban armas químicas. Numerosos líderes internacionales, entre ellos el presidente estadounidense, Donald Trump, acusan al Gobierno sirio de los hechos, y antes de que se lleve a cabo investigación alguna, Donald Trump ordena el bombardeo, el 7 de abril de 2017, de la base de Sharyat mediante el lanzamiento desde buques estadounidenses de 59 misiles de crucero Tomahawk. Antes del bombardeo advierte a Rusia del ataque, y esto permite retirar algunos aviones de la zona, pero aún así, destruyen de nueve a veinte aviones, y fallecen casi una decena de soldados sirios.
No contentos con el éxito conseguido con el ataque químico en Khan Shaykhun, el 7 de abril de 2018 tiene lugar otro incidente químico, supuestamente con una mezcla de cloro y sarín, esta vez, en Douma. El ataque dejó como saldo 50 personas muertas y alrededor de 500 heridos. Según la Organización de Voluntarios de la Defensa Civil Siria (pro-oposición siria) el ataque lo realizó el gobierno del presidente Bashar al-Asad para eliminar a los remanentes rebeldes, y lograr la conquista definitiva de Ghouta oriental. Sin esperar a investigación alguna, el 14 de abril de 2018, Estados Unidos, Reino Unido, y Francia bombardean objetivos que se suponen instalaciones de armas químicas del gobierno sirio.
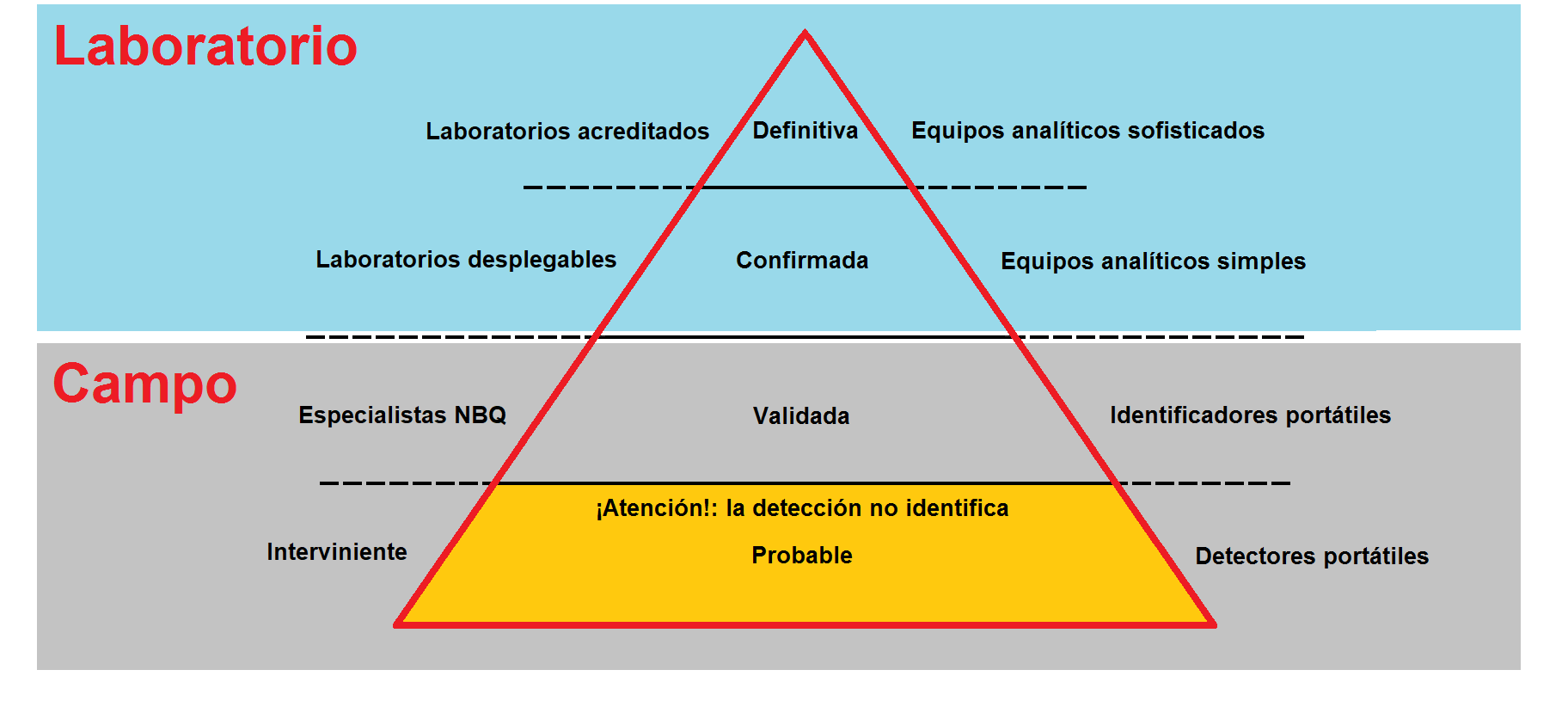
El 4 de mayo de 2018, la OPAQ informa que el despliegue inicial de la Misión de Investigación de los Hechos (FFM) en Douma, se ha completado y que las muestras tomadas han sido remitidas al Laboratorio de la OPAQ, donde una vez divididas serán enviadas a los Laboratorios acreditados para su análisis, que se estima tardarán por lo menos tres ó cuatro semanas. Hasta la fecha nada se sabe de estos análisis, ni del informe correspondiente de la Misión de Investigación de los Hechos21.
Referencias:
- «Armas químicas: la ciencia en manos del mal», René Pita, Plaza y Valdés Editores, 2008
- «NATO glossary of terms and definitions (english and french)», AAP-06, Edition 2015, https://www.unap.ro/ro/news/aap6.pdf
- «Convención sobre la prohibición del desarrollo, la producción, el almacenamiento y el empleo de armas químicas, y sobre su destrucción», https://www.opcw.org/fileadmin/OPCW/CWC/CWC_es.pdf
- «Chemical Warfare in World War I: The American Experience, 1917-1918», Charles E. Heller, Combat Studies Institute, Leavenworth Papers, 1984
- «Steel Wind: Colonel Georg Bruchmuller and the Birth of Modern Artillery», David T. Zabecki, Praeger, 1994
- «The Chemical Warfare Service: from laboratory to field», L. P. Brophy, W. D. Miles & R. C. Cochrane, Center of Military History, United States Army, 1959.
- «The problem of chemical and biological warfare, Volume 2: CB weapons today», Stockholm International PEACE Research Institute (SIPRI), Estocolmo,1973
- «Chemical Weapons and the Iran-Iraq War:A Case Study in Noncompliance», Javed Ali, The Nonproliferation Review, 2001, vol. 8, n.º 1
- «A poisonous affair: America, Iraq, and the gassing of Halabja», Joost R. Hiltermann, Cambridge University Press, 2007.
- «Tres tipos de inspecciones», Ficha descriptiva nº 5, OPAQ, https://www.opcw.org/fileadmin/OPCW/Fact_Sheets/Spanish/Fact_Sheet_5_-_Inspections.pdf
- «¿Completada la destrucción de las armas químicas sirias?», J. Domingo, https://cbrn.es/?p=433
- «Use of chemical weapons in the Syrian Civil War», Wikipedia, https://en.wikipedia.org/wiki/Use_of_chemical_weapons_in_the_Syrian_Civil_War
- «Timeline of Syrian Chemical Weapons Activity, 2012-2018», The Arms Control Association, https://www.armscontrol.org/factsheets/Timeline-of-Syrian-Chemical-Weapons-Activity
- «United Nations mission to investigate allegations of the use of chemical weapons in the Syrian Arab Republic» https://unoda-web.s3.amazonaws.com/wp-content/uploads/2015/01/UN-Mission-Syrian-Chemical-Weapons-Fact-Sheet-Jan2015.pdf
- «Informe de la Misión de las Naciones Unidas para Investigar las Denuncias de Empleo de Armas Químicas en la República Árabe Siria sobre el presunto empleo de armas químicas en la zona de Ghouta (Damasco) el 21 de agosto de 2013», Naciones Unidas, A/67/997–S/2013/553, http://www.un.org/es/comun/docs/?symbol=S/2013/553
- «Informe final de la Misión de las Naciones Unidas para Investigar las Denuncias de Empleo de Armas Químicas en la República Árabe Siria», Naciones Unidas, A/68/663–S/2013/735, http://www.iri.edu.ar/images/Documentos/Boletines_IRI/139/ONU_informe_final_sobre_siria.pdf
- «Decisión (PESC) 2017/2303 del Consejo, de 12 de diciembre de 2017, de apoyo a la aplicación continua de la Resolución 2118 (2013) del Consejo de Seguridad de las Naciones Unidas y la Decisión EC-M-33/DEC.1 del Consejo Ejecutivo de la Organización para la Prohibición de las Armas Químicas sobre la destrucción de las armas químicas sirias, en el marco de la aplicación de la Estrategia de la UE contra la proliferación de armas de destrucción masiva», https://eur-lex.europa.eu/legal-content/ES/TXT/PDF/?uri=CELEX:32017D2303&from=EN
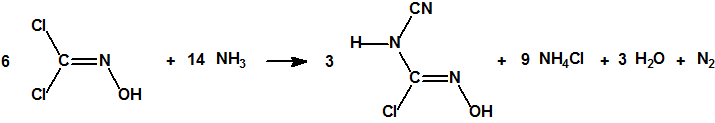
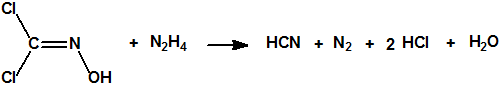
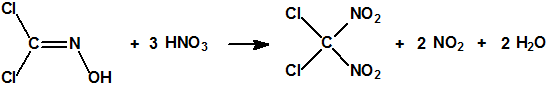
- «Sobre el cloro como método de guerra», J.Domingo, https://cbrn.es/?p=10
- «Resolución 2235 (2015) acerca de la creación de un mecanismo conjunto de investigación de la OPAQ y las Naciones Unidas», Unidas, S/RES/2235 (2015), http://undocs.org/es/S/RES/2235(2015)
- S/1510/2017 de fecha 29 de junio de 2017, «REPORT OF THE OPCW FACT-FINDING MISSION IN SYRIA REGARDING AN ALLEGED INCIDENT IN KHAN SHAYKHUN, SYRIAN ARAB REPUBLIC APRIL 2017», https://www.opcw.org/fileadmin/OPCW/Fact_Finding_Mission/s-1510-2017_e_.pdf
- «OPCW Spokesperson’s Statement on Fact-Finding Mission Deployment to Douma», OPCW, https://www.opcw.org/news/article/opcw-spokespersons-statement-on-fact-finding-mission-deployment-to-douma/