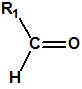
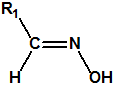
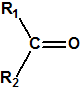
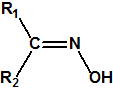
Las oximas son un grupo de compuestos orgánicos de fórmula general R1R2C=NOH, donde R1 es una cadena orgánica y R2 puede ser un hidrógeno o una cadena orgánica. La palabra oxima parece que proviene de la contracción de las palabras oxígeno e imina; las iminas son compuestos orgánicos, con estructura general R1R2C=NR3, donde R3 puede ser un H o una cadena orgánica, producto de condensación del amoníaco o de una amina primaria con una cetona o un aldehído. Las oximas cuando provienen de la condensación de la hidroxilamina con un aldehído, se denominan aldoximas, mientras que si provienen de la condensación con una cetona se denominan cetoximas1:
 |
 |
 |
 |
| Aldehido | Aldoxima | Cetona | Cetoxima |
Al igual que el doble enlace de los alquenos, el doble enlace de las oximas puede presentar isomería cis-trans (Z/E) cuando los sustituyentes R1 y R2 son diferentes. La estabilidad relativa de un isómero respecto del otro es de esperar que siga los mismos criterios que para los alquenos.
Las oximas pueden prepararse por condensación de un aldehído o de una cetona con hidroxilamina:
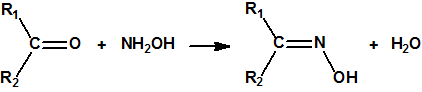
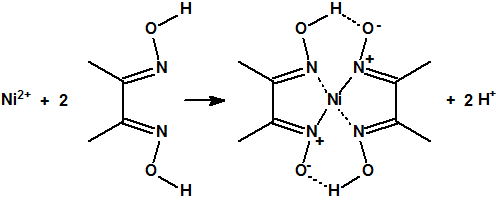
Las oximas son habitualmente sólidos cristalinos, que antes de la aparición de los métodos espectroscópicos, se utilizaban para separar y caracterizar compuestos con el grupo carbonilo. Las oximas también se utilizan como agentes formadores de complejos en algunas extracciones metálicas o para la determinación de ciertos iones metálicos, por ejemplo, la dimetilglioxima se utiliza para la determinación gravimétrica del Ni2+:
En el ámbito NBQ, determinadas oximas se utilizan como antídotos para las intoxicaciones con agentes neurotóxicos. Los agentes neurotóxicos inactivan la acetilcolinesterasa por fosforilación y ciertas oximas pueden reactivar la acetilcolinesterasa uniéndose al átomo de fósforo para formar el fosfo-derivado correspondiente que deja libre la molécula de acetilcolinesterasa:
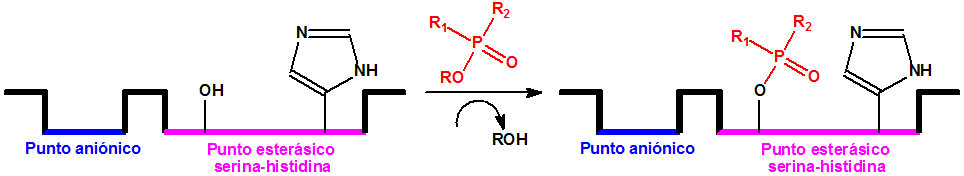
Inactivación de la acetilcolinesterasa por fosforilación

Reactivación de la acetilcolinesterasa por acción de la oxima, con liberación del fosfoderivado correspondiente
Entre las oximas empleadas como antídotos frente a los agentes neurotóxicos podemos citar la pralidoxima (también conocida como 2-PAM), la obidoxima, la metoxima, la HI-6, la HLö-7 y la TMB-4:
 |
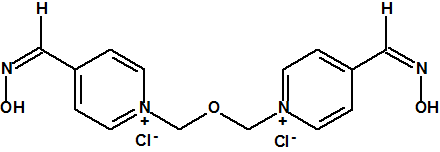 |
| Cloruro de pralidoxima, CAS 51-15-0 | Cloruro de obidoxima, CAS 114-90-9 |
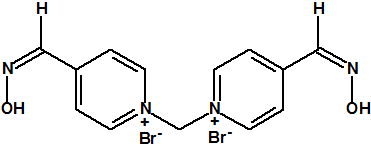 |
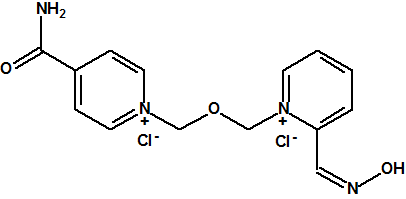 |
| Bromuro de metoxima, CAS 2058-89-1 | Cloruro de asoxima (HI-6), CAS 34433-31-3 |
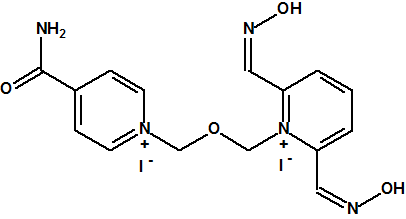 |
 |
| Yoduro de HLö-7, CAS 120103-35-7 | Bromuro de trimedoxima (TMB-4), CAS 56-97-3 |
Sin embargo la oxima del fosgeno no tiene utilidad industrial ni tampoco analítica, y es considerada un agente químico de guerra.
La oxima del fosgeno es una oxima mala.
La oxima del fosgeno2,3,4,5,6,7
La oxima del fosgeno, de fórmula empírica CHNCl2O y peso molecular 113,93, es la oxima del dicloruro de carbonilo, con número CAS 1794-86-1, y estructura química:
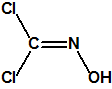
También se conoce como dicloroformoxima, 1,2-dichloroformoxima, dicloroformaldoxima, dicloroximinometano, dicloroformaldehido oxima, diclorometilen-hidroxilamina y CK (acrónimo militar). La oxima del fosgeno no está incluida en ninguna de las tres Listas de la CAQ, ni tampoco está recogida en el Grupo Australia.
La dicloroformoxima forma cristales prismáticos, incoloros y delicuescentes, que funden entre 39 °C y 40 °C. Incluso a las temperaturas ordinarias presenta una presión de vapor bastante alta. Tiene un punto de ebullición de 129 °C (con descomposición si no está muy pura), y a 28 mmHg de presión hierve a 53-54° C. Sus vapores tienen un olor penetrante y desagradable, y son más densos que el aire (drel=3,9). A 50 °C presenta una presión de vapor de 2,43 x 101 torr y una volatilidad estimada de 1,37 x 105 mg/m3.
La dicloroformoxima fue preparada en 1929 por los químicos alemanes Wilhelm Prandtl y Kurt Sennewald mediante la reducción del tricloronitrosometano (CAS 3711-49-7) con sulfuro de hidrógeno (CAS 7783-06-4)8:

Concluida la reacción, la dicloroformoxima se lava con agua, se filtra para eliminar el azufre, se seca con cloruro cálcico, se extrae con éter y se destila a vacío.
En vez de sulfuro de hidrógeno que es un gas tóxico, puede utilizarse como reductor una amalgama de aluminio. También puede prepararse por cloración del fulminato de mercurio, con posterior extracción con éter y destilación a vacío, lográndose un rendimiento del 65%:
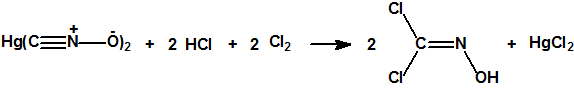
La patente «Process for preparing phosgene oxime», United States Patent 2299742, Philip J. Ehman and Walter O. Walker, Oct. 27, 1942, describe la obtención de la oxima del fosgeno por cloración de una solución acuosa de cloroisonitrosoacetona9:
La patente «Electrolytic production of dichloroformoxime», United States Patent 2918418, John H. Madaus & Herman B. Urbach, Dec. 22, 1959, describe la producción de dicloroformoxima por la reducción electrolítica de cloropicrina en un electrolito de ácido sulfúrico-alcohol, seguido de la recuperación de la dicloroformoxima mediante un procedimiento de extracción con cloropicrina10.
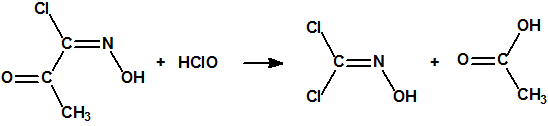
El procedimiento de obtención más habitual, muy sencillo pero algo más costoso, se basa en la reducción de la cloropicrina con ácido clorhídrico y estaño, que produce dicloroformoxima con una pureza del 85%. La reacción se lleva a cabo a 0 °C empleando tetrahidrofurano como disolvente. Al cabo de unas 6 horas, finalizada la reacción, se filtra para eliminar las sales insolubles de estaño, se evapora el tetrahidrofurano y el residuo se destila a vacío un par de veces11:
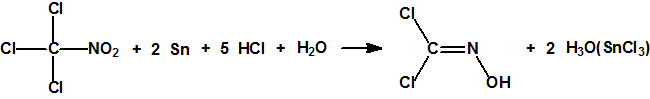
El procedimiento es similar al descrito en la patente «Production of dichloroformoxime», United States Patent US4558160, William R. Hydro, Dec. 10,198512.
La dicloroformoxima es una sustancia relativamente estable, soluble en agua y en los disolventes orgánicos más comunes. En solución acuosa sufre una hidrólisis lenta, según la reacción:
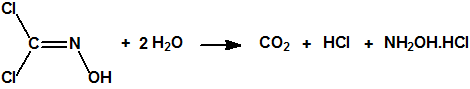
En presencia de ácidos diluidos la velocidad de hidrólisis aumenta y la hidrólisis es cuantitativa.
Los hidróxidos alcalinos y los carbonatos reaccionan enérgicamente con las soluciones acuosas de dicloroformoxima, con desprendimiento de calor, mientras la solución se vuelve amarilla.
Por la acción del amoníaco acuoso sobre una solución etérea de dicloroformoxima, se forma cianamido cloroformoxima junto con otros productos, según la reacción:
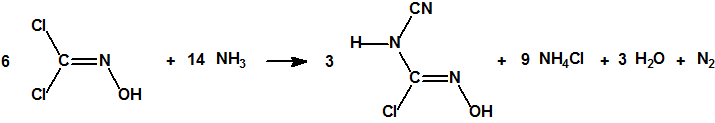
La cianamido cloroformoxima, que forma cristales incoloros que funden a 168° C, no tiene poder vesicante alguno.
Por la acción de la hidracina sobre una solución acuosa de dicloroformoxima, se forma ácido cianhídrico según la siguiente reacción:
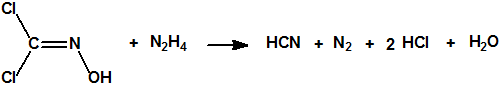
Con ácido nítrico fumante se transforma en diclorodinitrometano (CAS 1587-41-3):
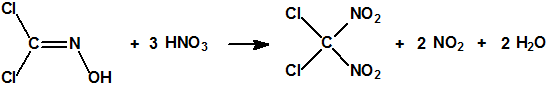
El diclorodinitrometano es un líquido de punto de ebullición 121,5 °C y densidad 1,872 g/mL, que explosiona si se intenta destilar a presión atmosférica, de modo que se destila a 31 °C/13 mbar (9,8 mmHg) (a 40 ºC a una presión de 12 mmHg).
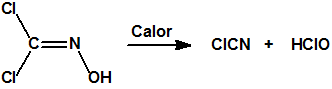
La dicloroformoxima sometida a calentamiento bajo reflujo, se descompone poco a poco en las proximidades de su punto de ebullición generando vapores de color marrón, de cloruro de cianógeno y ácido hipocloroso:
La dicloroformoxima, incluso cuando se almacena en recipientes sellados de vidrio o de cuarzo, se descompone a temperaturas ordinarias con formación de fosgeno y separación de un compuesto líquido. La descomposición es prácticamente completa en 3-4 semanas, pero está influenciada por la humedad y la temperatura. Los vapores de dicloroformoxima atacan el caucho y el corcho.
Aunque la oxima del fosgeno puede estimarse mediante técnicas colorimétricas y mediante pirolisis-cromatografía de gases, no existen apenas detectores portátiles que permitan su detección en un incidente. Un detector como el AP4C, que emplea la técnica de fotometría de llama (y detecta azufre, fósforo, arsénico y nitrógeno), si podría detectar la presencia de nitrógeno.
Toxicidad6,13,14,15,16
Aunque la oxima del fosgeno (dicloroformoxima o CX) es considerada un agente vesicante, no forma ampollas como los agentes vesicantes clásicos (mostazas de azufre, lewisitas y mostazas de nitrógeno), de modo que se considera también como un agente urticante, irritante o corrosivo, frente al cual no hay antídoto específico disponible. En términos coloquiales, los agentes vesicantes son sustancias químicas que provocan la aparición de ampollas en los tejidos afectados.
Las oximas halogenadas: diiodoformoxima, dibromoformoxima, monocloroformoxima y dicloroformoxima, fueron sintetizadas a finales de la década de 1920. La dicloroformoxima es la más irritante y se conoce comúnmente como la oxima del fosgeno. La oxima de fosgeno aunque sintetizada en 1929, muy probablemente nunca se haya utilizado en el campo de batalla. La oxima del fosgeno es uno de los agentes químicos de guerra menos conocido, y también uno de los menos estudiados.
No existen datos toxicológicos experimentales en humanos, pero la DL50 estimada para la oxima de fosgeno por contacto con la piel es de 25 mg×kg-1. Probablemente la oxima de fosgeno no tiene efectos directos adversos sobre la función reproductiva. El riesgo de exposición depende de lo cerca que se encuentren los individuos al lugar donde se haya liberado la oxima de fosgeno. Si se liberan vapores de la oxima de fosgeno existirá riesgo de exposición tanto a la inhalación de los vapores como al contacto de los mismos con la piel o con los ojos.
La oxima del fosgeno en forma de líquido y de vapor provoca, en contacto con ojos, piel y membranas mucosas, un dolor intenso y la destrucción local de los tejidos. Como ya se ha indicado no produce ampollas pero suele incluirse junto a los agentes vesicantes porque produce en los ojos, en los pulmones y en la piel daños similares a los producidos por los agentes vesicantes clásicos. En concentraciones inferiores al 8% hace poco daño biológico. En concentraciones más altas, sin embargo, causa daños más graves que cualquier otro vesicante, y las lesiones son similares a los causadas por la iperita o «gas mostaza». Provoca de manera instantánea un dolor tan intenso que los afectados tratan de quitarse las ropas o el equipo de protección para aliviar de algún modo el dolor producido. Al dolor producido tras la exposición le sigue una rápida necrosis de los tejidos.
Aunque tras la exposición a la oxima del fosgeno, el dolor inmediato advierte de la necesidad de utilizar equipo de protección y de proceder a la descontaminación, concentraciones bajas producen en los ojos lagrimeo y daños importantes, con inflamación y ceguera temporal, mientras que concentraciones altas pueden provocar ceguera y lesiones corneales permanentes. Al igual que los agentes vesicantes clásicos también aparece conjuntivitis, blefaritis, blefaroespasmo, lagrimeo y queratitis.
Las lesiones cutáneas son eritomatosas y extremadamente dolorosas. La irritación cutánea es inmediata y se asemeja a la producida por las ortigas. El contacto breve con sólo unos pocos miligramos produce dolor y picazón muy intensos. En menos de un minuto el área expuesta se vuelve blanca y queda rodeada por una zona eritomatosa circular que se asemeja a una diana, momento en el que la oxima del fosgeno es absorbida completamente por la piel. En menos de una hora la zona se convierte en edematosa, y en el plazo de 24 horas aparece el edema, la lesión se pigmenta de un color más oscuro, y aparece necrosis grave. Aparece descamación con la necrosis de la piel con formación durante los 7-10 días siguientes de una costra purulenta. La lesión necrótica acaba extendiéndose al panículo y al músculo, rodeada por una inflamación intensa.
La urticaria provocada en la piel por la oxima del fosgeno se asemeja a la causada por reacciones alérgicas y no alérgicas a diferentes sustancias ambientales y se cree que puede ser debida principalmente a la activación de los mastocitos y a la liberación de histamina. Aunque se desconoce el mecanismo de acción se ha sugerido que probablemente posea propiedades alquilantes y nucleofílicas semejantes a las de los agentes vesicantes clásicos, y por lo tanto sus efectos pueden ser directos, con lesiones corrosivas, muerte celular y destrucción de tejidos, e indirectos, relacionados con las células inflamatorias como los mastocitos y los neutrófilos que provocan lesiones tardías en los tejidos.
La exposición por inhalación puede causar irritación inmediata del tracto respiratorio, disnea e incluso edema pulmonar, pues la absorción es completa en segundos. El edema pulmonar puede venir acompañado por bronquiolitis necrotizante y por trombosis venosa pulmonar. La exposición a 0,2 mg×min×m-3 produce irritación, que resulta intolerable a 3 mg×min×m-3. La CLt50 estimada es de 1500-2000 mg×min×m-3. La intoxicación por vía oral es muy similar en curso a la intoxicación por vía inhalatoria.
No hay tratamiento específico disponible para lesiones producidas por la oxima del fosgeno. Los afectados deberían ser trasladados inmediatamente a zona limpia para así reducir la exposición, y puesto que los vapores son más densos que el aire, las zonas más altas son las más apropiadas. El objetivo de la terapia será aliviar los síntomas, prevenir las infecciones y promover la curación. En casos de ingestión oral se recomienda la dilución con agua o leche. Debido a los efectos irritantes y corrosivos de la oxima del fosgeno no se recomiendan ni el vómito (emesis) ni el empleo de carbón activo. Las lesiones necróticas de la piel deben tratarse quirúrgicamente, y el edema pulmonar tratarse apropiadamente. La recuperación total tarda de 1 a 3 meses, pero algunas quemaduras pueden tardar más de 6 meses en sanar.
Los ojos deben enjuagarse con abundante agua tibia hasta que los lixiviados tengan pH neutro. La descontaminación de los ojos debe ser inmediata pues la oxima del fosgeno oxima se absorbe en cuestión de segundos. Las úlceras corneales deben tratarse atropina oftálmica para prevenir daños mayores. No se recomienda el uso de anestésicos tópicos para aliviar el dolor, ya que pueden aumentar el daño corneal. Por el contrario, la ausencia de luz (oscuridad) y el uso sistémico de analgésicos opiodes pueden resultar beneficiosos.
Descontaminación3,6
La descontaminación de la piel se basa en la adsorción física o en la combinación de adsorción física y de inactivación química. La adsorción física se consigue con polvos adsorbentes, por ejemplo, polvo de talco, o tierra de fullers (arcilla a base de silicatos de aluminio hidratados), mientras que la inactivación química se consigue por la acción de sustancias alcalinas. Los agentes clorados como la lejía no funcionan con fosgeno oxima. La descontaminación de los agentes vesicantes no debería realizarse con agua, excepto los ojos, ya que con ello puede producirse la diseminar del agente. La descontaminación cutánea debe llevarse a cabo inmediatamente, ya que la absorción total por la piel se produce en cuestión de minutos. La oxima del fosgeno reacciona rápidamente con el tejido y una vez que aparece el dolor la descontaminación no resulta eficaz (10).
Las sustancias utilizadas para la descontaminación cutánea suelen ser demasiado irritantes para su uso en los ojos, de modo que los ojos deben ser enjuagados inmediatamente con copiosas cantidades de agua o bicarbonato sódico isotónico (solución acuosa de hidrogeno carbonato sódico al 1,26%).
La ropa contaminada con oxima del fosgeno supone un peligro inmediato, por lo que se recomienda su retirada inmediata, y su colocación en una bolsa de plástico que debe cerrarse convenientemente para evitar la salida de los vapores.
Referencias
- «IUPAC Gold Book-Oximes», http://goldbook.iupac.org/html/O/O04372.html
- «Potential military CB agents and compounds», FM 3-11.9, 2005, https://fas.org/irp/doddir/army/fm3-11-9.pdf
- «Handbook of Chemical and Biological Warfare Agents», D. Hank Ellison, CRC Press, 2Ed., 2007
- «Compendium of Chemical Warfare Agents», Steven L. Hoenig, Springer, 2007
- «The war gases», Mario Sartori, D. Van Nostrand Co., 1939
- «Phosgene oxime-forgoten chemical weapon», Jiří Patočka & Kamil Kuča, Mil. Med. Sci. Lett. (Voj. Zdrav. Listy) 2011, vol. 80, p. 38-41.
- «A Review of the Scientific Literature as it Pertains to Gulf War Illnesses», Volume 5: Chemical and Biological Warfare Agents, Chapter Three: «Skin-Damaging Agents», William Augerson, RAND Corporation, 2000, https://www.rand.org/pubs/monograph_reports/MR1018z5.html
- «Trichloronitrosomethane, Dichloroformoxime (Phosgene Oxime) and Their Derivatives», Wilhelm Prandtl & Kurt Sennewald, Chemische Berichte, Vol. 62, p. 1766, 1929.
- «Process for preparing phosgene oxime», United States Patent 2299742, Philip J. Ehman and Walter O. Walker, Oct. 27, 1942.
- «Electrolytic production of dichloroformoxime», United States Patent 2918418, John H. Madaus & Herman B. Urbach, Dec. 22, 1959
- «A Laboratory History of Chemical Warfare Agents», Jared Ledgard, 2Ed., 2006
- «Production of dichloroformoxime», United States Patent US4558160, William R. Hydro, Dec. 10,1985.
- «A Toxico-Pathologic Study of Phosgene Oxime», Arthur J.McAdams, & Milton H. Joffe, Medical Laboratories Research Report No. 381, July 1955.
- «Handbook of Toxicology of Chemical Warfare Agents», Ramesh C. Gupta, Elsevier,2ªEd., 2015
- «Cutaneous exposure to vesicant phosgene oxime-Acute effects on the skin and systemic toxicity», N. Tewari-Singh, D. G. Goswami, R. Kant, C. R. Croutch, R. P. Casillas, D. J. Orlicky & R. Agarwal, Toxicology and Applied Pharmacology 317 (2017) 25–32
- «Phosgene oxime: Injury and associated mechanisms compared to vesicating agents sulfur mustard and lewisite «, G. Goswami, R. Agarwal & N. Tewari-Singh, Toxicology letters, 2017