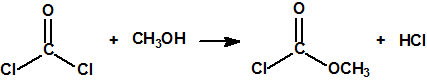El día 11 de marzo de 2011 (año del conejo) tuvo lugar el terremoto y posterior tsunami, conocido como el terremoto de la costa del Pacífico en la región de Tohoku o Gran terremoto de Japón oriental, que originó un grave accidente en la planta nuclear de Fukushima Daiichi. Así pues, hoy 11 de marzo de 2017 (año del gallo) habrán transcurrido seis años desde el accidente, y los problemas de Fukushima Daiichi aún no se han resuelto completamente.
Planta nuclear de Fukushima Daiichi
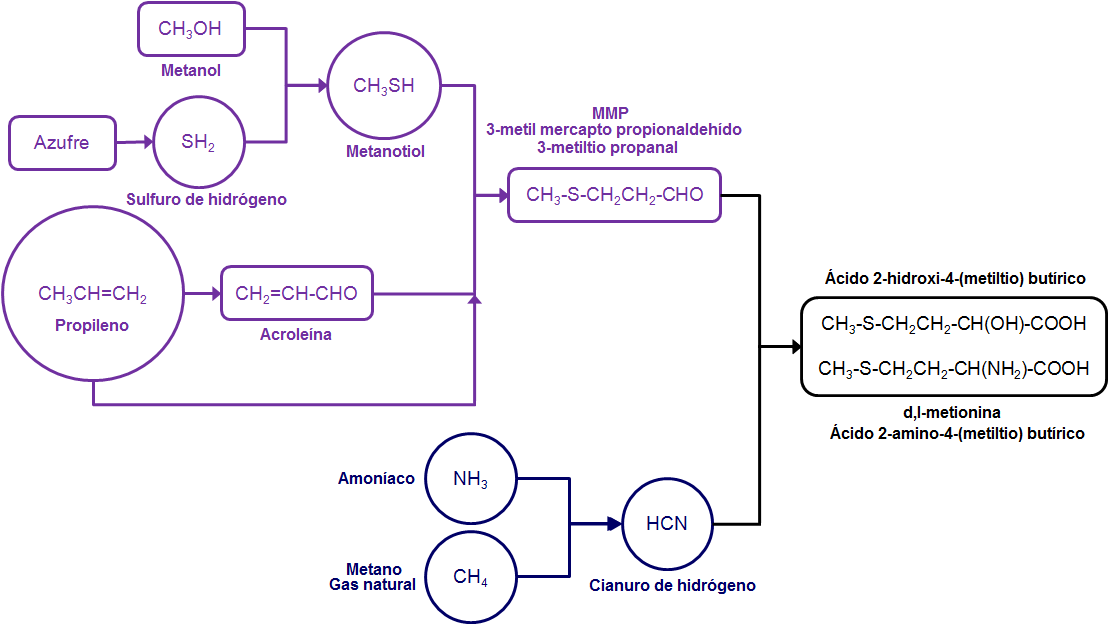
La central de nuclear de Fukushima Daiichi o Fukushima -I albergaba seis reactores de agua de ebullición (BWR, Boiling Water Reactor). Fukushima Daiichi es la primera estación de energía nuclear de Tokyo Electric Power Company (TEPCO). El reactor nº1 entró en operación en marzo de 1971, y los demás fueron agregados paulatinamente, hasta la entrada en operación del reactor nº 6 en octubre de 1979. La capacidad total de generación de potencia instalada es de 4696 millones de kW.1
Los vasos de contención (CV) de los reactores son de los tipos BWR-3 y ABWR . Los tipos BWR-3 y BWR-4 son Mark I con una piscina de supresión en forma de donut, mientras que el tipo BWR-5 (ABWR) es Mark II y ya no tiene esa forma de donut. La tabla siguiente muestra un resumen de las características de los reactores de Fukushima Daiichi. 1,2
| Reactor nº1 | Reactor nº2 | Reactor nº3 | Reactor nº4 | Reactor nº5 | Reactor nº6 | |
| Potencia (MWe) | 460 | 784 | 784 | 784 | 784 | 1,100 |
| Potencia térmica (MWt) | 1380 | 2381 | 2381 | 2381 | 2381 | 3293 |
| Puesta en operación | 3/1971 | 7/1974 | 3/1976 | 10/ 1978 | 4/1978 | 10/ 1979 |
| Tipo de reactor nuclear | BWR-3 | BWR-4 | BWR-4 | BWR-4 | BWR-4 | BWR-5 |
| Fabricante | GE | GE-Toshiba | Toshiba | Hitachi | Toshiba | GE-Toshiba |
| Presión de diseño para la vasija de presión del reactor (kg/cm2) | 87,9 | 87,9 | 87,9 | 87,9 | 87,9 | 87,9 |
| Temperatura de diseño para la vasija de presión del reactor ( °C) | 302 | 302 | 302 | 302 | 302 | 302 |
| Número de elementos de combustible | 400 | 548 | 548 | 548 | 548 | 764 |
| Numero de barras de control | 97 | 137 | 137 | 137 | 137 | 185 |
| Tipo de vasija de contención | Mark I | Mark I | Mark I | Mark I | Mark I | Mark II |
| Presión de diseño para la vasija de contención (kg/cm2) | 4,35 | 3,92 | 3,92 | 3,92 | 3,92 | 2,85 |
| Temperatura de diseño para la vasija de contención ( °C) | 138 (D/W) | 138 (D/W) | 138 (D/W) | 138 (D/W) | 138 (D/W) | 171 (D/W) |
| 138 (S/C) | 138 (S/C) | 138 (S/C) | 138 (S/C) | 138 (S/C) | 105 (S/C) | |
| Configuración del sistema de refrigeración de emergencia del núcleo, ECCS
|
HPCI | HPCI | HPCI | HPCI | HPCI | HPCS |
| CS | CS | CS | CS | CS | LPCS | |
| ADS | LPCI | LPCI | LPCI | LPCI | LPCI | |
| ADS | ADS | ADS | ADS | ADS | ||
| Sistema de refrigeración de aislamiento del núcleo del reactor , RCIC | IC | RCIC | RCIC | RCIC | RCIC | RCIC |
| Significado de las abreviaturas:
(ECCS, Emergency Core Cooling System), Sistema de refrigeración de emergencia del núcleo (CS, Core Spray), Aspersión del núcleo (HPCI, High Pressure Coolant Injection), Inyección de refrigerante a alta presión (LPCI, Low Pressure Coolant Injection), Inyección de refrigerante a baja presión (ADS, Automatic Depressurization System), Sistema automático de despresurización (IC, Isolation Condenser ), Condensador de aislamiento (RCIC, Reactor core Isolation Cooling), Refrigeración de aislamiento del núcleo del reactor |
||||||
| |
|
| Mark I (imagen de http://nuclearstreet.com/) |
Mark II (imagen de http://nuclearstreet.com/) |
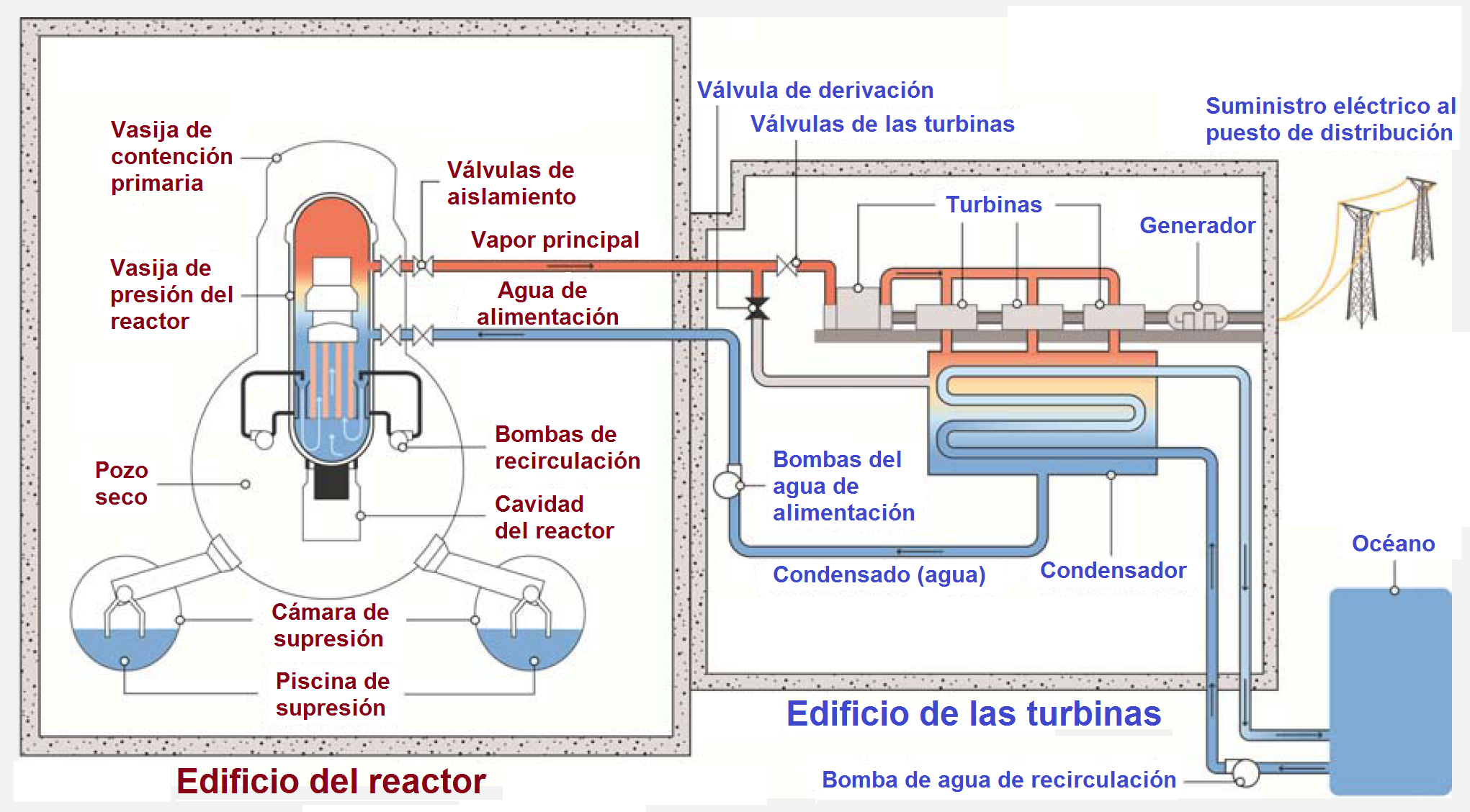
Esquema de un BWR Mark I, adaptado de «The Fukushima Daiichi Accident, Technical Volume 1/5, Description and Context of the Accident», IAEA, 2015, http://www-pub.iaea.org/MTCD/Publications/PDF/AdditionalVolumes/P1710/Pub1710-TV1-Web.pdf
El accidente nuclear de Fukushima Daiichi 3, 4, 5
El 11 de marzo de 2011 a las 14:46 (hora local en Japón), un fuerte terremoto de magnitud de 9.0 tuvo lugar en la región de Tohoku. El epicentro fue situado a 130 kilómetros al este de la ciudad de Sendai y a 163 km al noreste de la planta nuclear de Fukushima Daiichi. Cuando se produjo el terremoto tres de los seis reactores, los reactores 1-3, estaban en operación mientras que los reactores 4-6 estaban parados para recarga de combustible y mantenimiento5.
Como consecuencia directa del terremoto, se produjo la parada automática de los reactores nº1, nº2 y nº3 (entró en funcionamiento el sistema de apagado automático del reactor mediante la introducción automática o manual de las barras control, proceso conocido como SCRAM, Safety Control Rod Axe Man). El terremoto provocó la pérdida de suministro eléctrico en las instalaciones y los generadores diesel de emergencia se activaron. Estas condiciones por si solas no representarían una pérdida severa de las funciones de seguridad.
Al terremoto inicial (14:46 h, magnitud 9,0) le siguieron hasta 10 réplicas de magnitud superior a 6 durante un período de aproximadamente 40 minutos (15:08 h, magnitud 7,4; 15:15 h, magnitud 7,9; 15:25 h, magnitud 7,7).
Debido al terremoto, las 7 líneas de suministro eléctrico a Fukushima Daiichi quedaron inutilizables debido a las roturas de las líneas y caídas de las torres. La central sufrió la pérdida de alimentación externa, es decir un apagón debido a los daños en las líneas de suministro eléctrico.
El terremoto generó un gran tsunami que afectó a una amplia zona costera de la isla de Honshu, desde la prefectura de Iwate, al norte, hasta la prefectura de Ibaraki, próxima a Tokio. Unos 45 minutos después del terremoto, aproximadamente a las 15:27 h, llegó a Fukushima Daiichi la primera ola, de aproximadamente 4 m de altura y unos 8 minutos después, a las 15:35 llegó una segunda ola de 13-14 metros que resultó devastadora.
Los efectos del tsunami son como los de una enorme marea alta y no se limitan sólo a los efectos mecánicos de las olas. Tras la llegada de las olas todo queda cubierto por una extensa masa de agua, barro y escombros diversos, especialmente las zonas más bajas.
Los sótanos, bajos y primeros pisos de las instalaciones de la planta quedaron completamente sumergidos en el agua, y por ende todos los equipos en ellos contenidos. Por ejemplo, 12 de los 13 generadores de diesel de emergencia, para uso en caso de un fallo en el suministro externo de energía, quedaron inservibles a causa de la inundación. Además la mayoría de los paneles de distribución de energía instalados en los sótanos, bajos y primeros pisos también quedaron inservibles al quedar bajo el nivel del agua. Incluso si el suministro externo de energía eléctrica no hubiese fallado o hubiese sido restablecido, no es seguro que hubiera podido restablecerse el funcionamiento de los equipos.
Al final, además de la pérdida del suministro externo de energía eléctrica, también se perdió la alimentación de emergencia y la fuente de corriente continua (baterías) de modo que la central nuclear perdió todo suministro de electricidad y sufrió un apagón total, lo que se denomina «apagón de la planta» («station blackout»). Este apagón total continuó durante aproximadamente 10 días, hasta que pudo de algún modo restablecerse el suministro eléctrico y comenzaron los trabajos de recuperación de la planta.
Daños en los diferentes reactores 3, 4, 5
Reactor 1
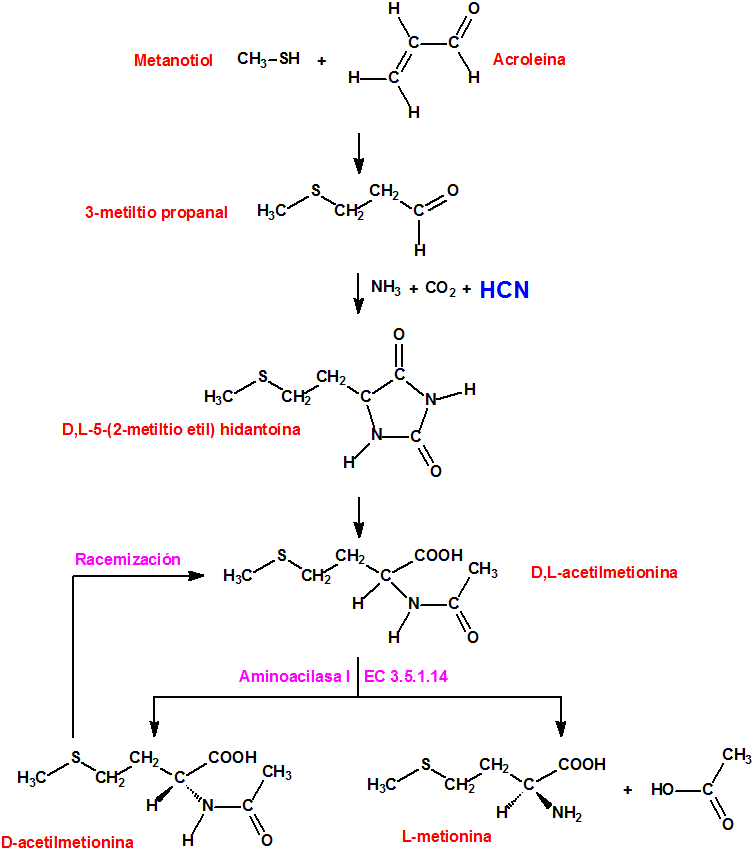
En la mañana del día 12 de marzo, la pérdida de refrigeración en el reactor condujo a un aumento de temperatura de las barras de combustible y a la reacción, a altas temperaturas, del circonio del revestimiento del combustible con el vapor de agua, para generar hidrógeno, y finalmente provocó la fusión de las barras de combustible.
Recordemos que en el caso de accidente de pérdida de refrigerante (LOCA) en un reactor nuclear, el revestimiento de circonio del combustible nuclear reacciona con el vapor de agua a alta temperatura para producir hidrógeno, en una reacción además exotérmica:
![]()
Debido a la formación de vapor de agua y de hidrógeno la presión en el interior de la vasija de contención aumentó y superó el valor límite de diseño (4,35 kg/cm2). A las 14:30 h del día 12 de marzo, para aliviar la presión, se abrió manualmente la válvula de alivio y seguridad, y se provocó una emisión (sin filtros) de material radiactivo.
Poco después, a las15:36 h, a pesar de la ventilación realizada, una explosión de hidrógeno en la parte superior del edificio del reactor, destruyó parte del mismo y provocó una nueva emisión de material radiactivo.
A las 20:20 h comenzó el enfriamiento del núcleo utilizando agua del mar, hasta que más tarde pudo sustituirse ésta con agua dulce para evitar así la erosión
Reactor 2
En el reactor nº2, el núcleo no pudo enfriarse adecuadamente debido a problemas de funcionamiento del sistema de refrigeración de aislamiento del núcleo del reactor (RCIC), que dejó de funcionar a las 13:25 h del día 14 de marzo. A las 19:57 h se inició la inyección de agua de mar, pero a pesar de todo parece que el día 15 de marzo hubo una ruptura debida a la presión, que provocó una nueva emisión de material radiactivo
Reactor 3
A las 11:36 h del día 12 de marzo, el sistema de refrigeración de aislamiento del núcleo del reactor (RCIC) del reactor nº3 se detuvo, y arrancó el sistema de inyección de refrigerante a alta presión (HPCI), que estuvo funcionando hasta que 2:42 h del día 13 de marzo se paró. A partir de ese momento la presión subió, se ventiló y a las 13:12 h del día 13 de marzo se empezó a bombear agua de mar. Pese a todo a las 11:01 h del día 14 de marzo, una explosión de hidrógeno en la vasija de contención destruyó el edificio.
Reactor 4
El reactor nº4 había sido apagado recientemente y el combustible movido a la piscina de combustible gastado (SFP, Spent Fuel Pool) que aún estaba caliente y necesitaba enfriamiento constante. A pesar de ello, parece que una fuga de hidrógeno procedente del reactor nº3, provocó, a las 6:14 h del día 15 de marzo, una explosión que dañó el edificio y dejó al descubierto, sin contención, algunas de las barras de combustible aún calientes.
Para una descripción más detallada de la secuencia de acontecimientos ocurridos en los diferentes reactores de Fukushima Daiichi puede consultarse la abundante bibliografía publicada al respecto. 1, 3, 4, 5, 6, 7, 8
Además, las barras de combustible nuclear gastado de los reactores nº1, nº2, nº3 y nº4, almacenadas en las piscinas de combustible comenzaron a sobrecalentarse cuando los niveles de dichas piscinas bajaron. El reactor nº3 representaba un mayor riesgo pues empleaba un combustible especialmente peligroso denominado «MOX», formado por una mezcla de uranio más plutonio10. «MOX», es la abreviatura de «Mixed OXide», «Mezcla de Óxidos». Se trata de un tipo de combustible constituido por una mezcla de óxido de uranio natural, uranio reprocesado o uranio empobrecido, y óxido de plutonio. La proporción de plutonio en este combustible varía de un 3% a un 10%, de modo que uno de los atractivos del «MOX» es que puede utilizarse para eliminar parte del plutonio de grado militar, eliminando el problema de su almacenamiento.
El miedo a filtraciones de radiación llevó a las autoridades a evacuar un radio de 20 km kilómetros alrededor de la planta, extendiendo luego este radio a 30 km y posteriormente a 40 km. Los trabajadores de la planta sufrieron exposición a radiación en varias ocasiones y fueron evacuados temporalmente en distintas ocasiones9,10.
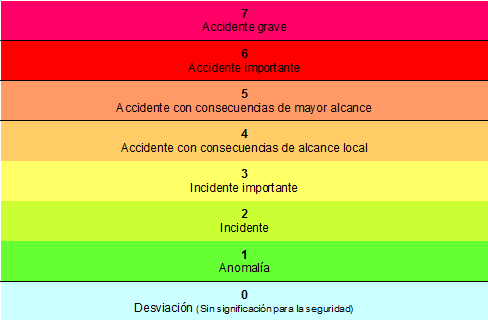
El accidente fue considerado inicialmente de nivel 4 en Escala Internacional de Eventos Nucleares (escala INES, por sus siglas en inglés). Aunque en los días siguientes la situación se agravó y el accidente nuclear acabó alcanzando el nivel 7, el mismo que el accidente, en 26 de abril de 1986, de la central nuclear de Chernobyl9.
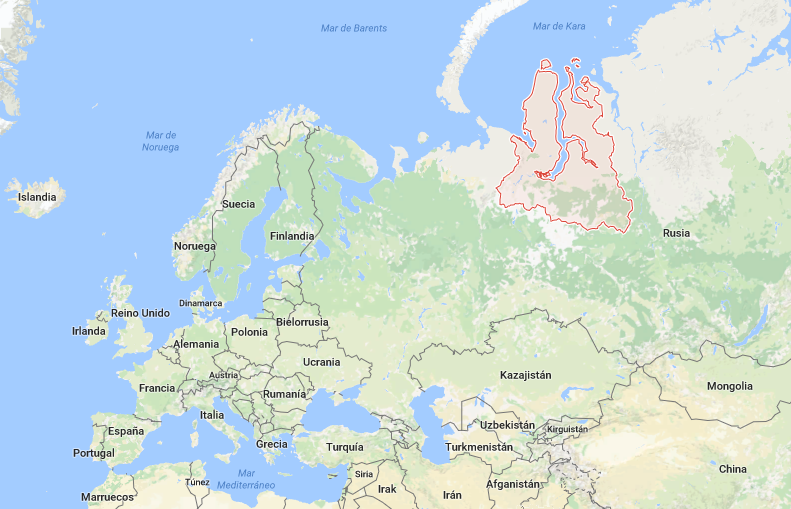
Tsunami para todos 5, 6
El terremoto y posterior tsunami golpeó 14 reactores de energía nuclear (11 de ellos estaban en operación) ubicados en 4 centrales nucleares situadas en la costa nororiental de la isla de Honshu, de cara al océano Pacífico del Japón:
- La central nuclear de Onagawa, de Tohoku Electric Power Co., con 3 reactores del tipo BWR,
- La central nuclear de Fukushima Daiichi o Fukushima-I, de Tokyo Electric Power Co. Ltd (TEPCO) con 6 reactores del tipo BWR,
- La central nuclear de Fukushima Daini o Fukushima-II, de Tokyo Electric Power Co. Ltd (TEPCO), con 4 reactores del tipo BWR, y
- La central nuclear de Tokai Daini, de Japan Atomic Power Company con un reactor nuclear del tipo BWR
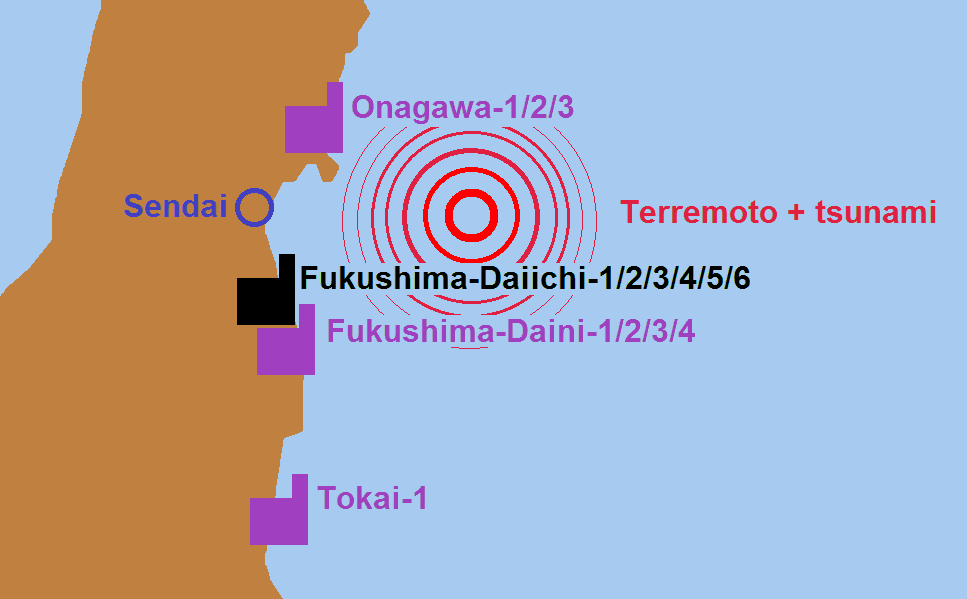
Esquema de la ubicación del terremoto+tsunami y de las plantas nucleares afectadas
A pesar de que todos los reactores nucleares de la costa oriental de la isla de Honshu, incluidos los reactores nº1, nº2 y nº3 de Fukushima Daiichi, resultaron ser sísmicamente robustos, el terremoto dañó seriamente las instalaciones de suministro eléctrico a las instalaciones.
Para estabilizar un reactor nuclear, es necesario alcanzar de manera segura la «parada fría» o «cool shutdown» utilizando el sistema de eliminación del calor residual (RHR, Residual Heat Removal). En el caso de los reactores en funcionamiento cuando sucedió el terremoto y posterior tsunami, en 8 de los 11 fue posible disponer de algún tipo suministro eléctrico para las bombas de los sistemas de eliminación del calor residual: los 3 reactores de Onagawa, los 4 reactores de Fukushima Daini y el reactor de Tokai. A pesar de los problemas habidos en algunos de ellos, en aproximadamente 4 días, fue posible alcanzar la «parada fría» en todos ellos (Cuando un reactor alcanza la «parada fría», la vasija de presión del reactor (RPV) puede abrirse con seguridad y con mucho cuidado para agregar agua a la cavidad como blindaje y permitir un manejo seguro del combustible, bien para reaprovisionar combustible (sustitución de elementos combustibles agotados) o bien para retirar el combustible).
Sólo los reactores nº1, nº2, nº3 y nº4 de Fukushima Daiichi tuvieron serios problemas debido sobre todo al tsunami. Los reactores de Fukushima Daiichi resultaron más vulnerables al tsunami debido a la altura que alcanzaron las olas, que llegó a ser más de dos veces superior a la altura prevista por TEPCO para un posible tsunami. La planta nuclear de Onagawa alcanzada por estas mismas olas no sufrió los mismos efectos al estar por su posición más protegida.
![]()
Esquema de las diferentes alturas en la planta de Fukushima Daiichi.Adaptada de «Fukushima Accident-Radioactivity Impact on the Environment», Pavel Povinec, Katsumi Hirose & Michio Aoyama, Elsevier, 2013
El Gobierno de Japón confirmó el 16 de diciembre de 2011 que los tres reactores nucleares de la central de Fukushima dañados por el tsunami habían han alcanzado la «parada fría», lo que supone que se mantienen de forma estable por debajo de 100 grados centígrados.11
Emisiones de material radiactivo1, 3, 12, 13
Las emisiones atmosféricas de radionucleidos durante el accidente de Fukushima se estima que han sido superiores a 153-160 PBq de 131I (período de semidesintegración de 8,02 días) y 13-15 PBq de 137Cs (período de semidesintegración de 30,2 años). Además de 131I y 137Cs, también se habrían emitido 132I (período de semidesintegración de 2,295 horas), 134Cs (período de semidesintegración de 2,06 años), 136Cs (período de semidesintegración de 13,16 días), 132Te (período de semidesintegración de 3,204 días) y gases nobles como 133Xe (período de semidesintegración de 5,247 días) y 135Xe (período de semidesintegración de 9,14 horas). (Un Peta bequerelio, PBq, equivale a 1015 bequerelios y un Tera bequerelio, TBq, equivale a 1012 bequerelios). Los isótopos del cesio por su periodo de semidesintegración, solubilidad y bioacumulación son sin duda los más peligrosos, pues a través de la cadena alimenticia, vegetales, animales, pescados y mariscos, llega a las personas.
El informe final de la Atomic Energy Society of Japan (Sociedad Japonesa de Energía Atómica) de 2015 menciona unas emisiones a la atmósfera de 500 PBq de 131I, 500 PBq de gases nobles, 10 PBq de 134Cs y 10 PBq de 137Cs. En cuanto a las emisiones al océano indica 11 PBq de 131I, 3,5 PBq de 134Cs y 3,6 PBq de 137Cs.
Los radionucleidos emitidos a la atmósfera sufren diferentes procesos físico-químicos y meteorológicos que llevan a su deposición más o menos rápida al terreno y a las aguas.
![]()
Modelo simple de los procesos atmosféricos que afectan a las emisiones de radionucleidos aerotransportados a raíz del accidente de Fukushima. Adaptado de «Radioiodine in the atmosphere after the Fukushima Daiichi nuclear accident», Luke S. Lebel, Raymond S. Dickson & Glenn A. Glowa, Journal of Environmental Radioactivity 151 (2016) 82-93
Hacia el desmantelamiento y la restauración14, 15
El 19 de julio de 2011, la Oficina de respuesta integrada Gobierno-TEPCO (Government–TEPCO Integrated Response Office) informó que se había alcanzado la condición de «dosis de radiación en declive», la primera de las condiciones establecidas para lograr el «estado de parada fría» necesario para la restauración.
Unos meses después, el 16 de diciembre de 2011, informó que se había logrado la segunda condición del «estado de parada fría» en los reactores nº1, nº2 y nº3 ya que: (1) la temperatura medida en la parte inferior de la vasija de presión del reactor (RPV) y la de los gases en la parte superior de la misma se mantenían por debajo de aproximadamente 100 °C; y (2) las dosis evaluadas de radiación debidas a la liberación de material radiactivo procedente de la vasija de presión del reactor serían del orden de 0,1 mSv/año, muy por debajo del objetivo de 1 mSv/año.
En base a esta declaración de consecución del «estado de parada fría» ese mismo día 16 de diciembre de 2011, se suprimió la Oficina de respuesta integrada Gobierno-TEPCO y se creó una nueva organización, el Consejo Gobierno -TEPCO de respuesta a medio y largo plazo (Government–TEPCO Mid-to-Long Term Response Council).
El 21 de diciembre de 2011, este Consejo hizo pública la hoja de ruta a medio y largo plazo para el desmantelamiento de los reactores nº1, nº2, nº3 y nº4 de Fukushima Daiichi.
El 18 de diciembre de 2013 se decidió desmantelar los reactores nº5 y nº6, con lo cual la planta entera de Fukushima Daiichi será desmantelada
El desmantelamiento y la recuperación de las zonas afectadas por la contaminación radiactiva tardarán decenas de años en conseguirse y tendrán un coste muy elevado, difícil de estimar incluso después de transcurridos seis años del accidente.
Referencias
- «The Fukushima Daiichi Nuclear Accident, Final Report of the AESJ Investigation Committee»,Atomic Energy Society of Japan, Springer, 2015
- «Insights from review and analysis of the Fukushima Dai-ichi accident», Masashi Hirano, Taisuke Yonomoto, Masahiro Ishigaki, Norio Watanabe, Yu Maruyama, Yasuteru Sibamoto, Tadashi Watanabe & Kiyofumi Moriyama, Journal of Nuclear Science and Technology, Volume 49, No. 1, January (2012) pp. 1–17
- «A Study of the Fukushima Daiichi Nuclear Accident Process-What caused the core melt and hydrogen explosión?», Michio Ishikawa, Springer, 2015
- «The Fukushima Daiichi nuclear accident-an overview», Harald Thielen, Health Physics August 2012, Volume 103, Number 2, 169-174
- «Fukushima accident: What happened?», M. Baba, Radiation Measurements 55:17-21, August 2013
- «Fukushima Accident-Radioactivity Impact on the Environment», Pavel Povinec, Katsumi Hirose & Michio Aoyama, Elsevier, 2013
- «Reflections on the Fukushima Daiichi Nuclear Accident», Joonhong Ahn, Cathryn Carson, Mikael Jensen, Kohta Juraku, Shinya Nagasaki & Satoru Tanaka, Spinger, 2015
- «The 2011 Fukushima Nuclear Power Plant Accident-How and Why it Happened», Yotaro Hatamura, Seiji Abe, Masao, Fuchigami & Naoto Kasahara,Elsevier, 2015
- «Accidente nuclear de Fukushima», https://energia-nuclear.net/accidentes-nucleares/fukushima.html
- «Accidente nuclear de Fukushima I», https://es.wikipedia.org/wiki/Accidente_nuclear_de_Fukushima_I
- «Los reactores nucleares de Fukushima, en «parada fría»», http://www.abc.es/20111216/internacional/abci-reactores-nucleares-fukushima-parada-201112160903.html
- «Fukushima Daiichi-Derived Radionuclides in the Ocean-Transport, Fate, and Impacts», Ken Buesseler, Minhan Dai, Michio Aoyama, Claudia Benitez-Nelson, Sabine Charmasson, Kathryn Higley, Vladimir Maderich, Pere Masqué, Deborah Oughton & John N. Smith, Annu. Rev. Mar. Sci. 2017. 9:1.1–1.31
- «Agricultural Implications of the Fukushima Nuclear Accident», Tomoko M. Nakanishi & Keitaro Tanoi, Springer, 2013
- «The Fukushima Daiichi Accident, Technical Volume 1/5, Description and Context of the Accident», IAEA, 2015, http://www-pub.iaea.org/MTCD/Publications/PDF/AdditionalVolumes/P1710/Pub1710-TV1-Web.pdf
- » Decommissioning of Units 5 and 6 at Fukushima Daiichi Nuclear Power Station», TEPCO, http://www.tepco.co.jp/en/announcements/2014/1233973_5932.html