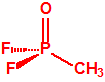
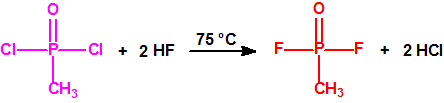
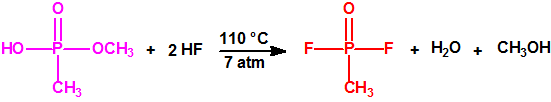
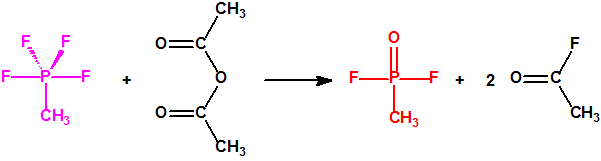
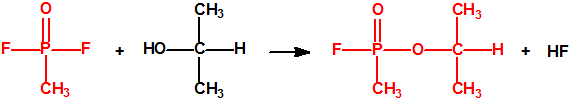
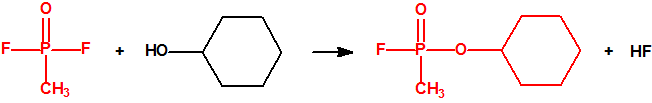
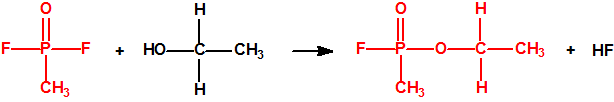
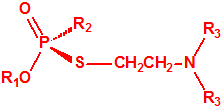
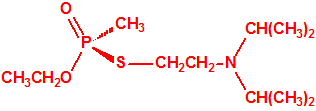
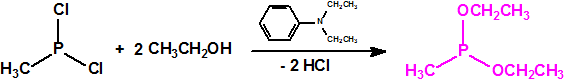
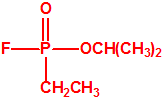
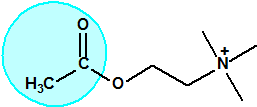
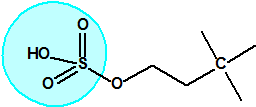
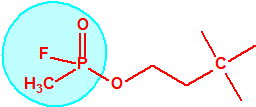
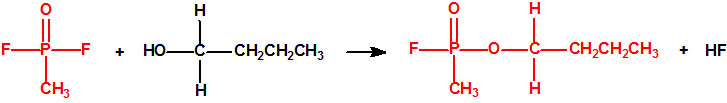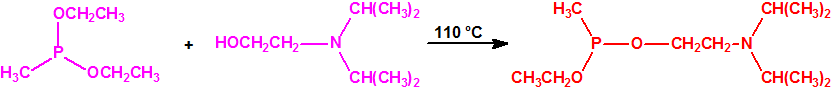La fosfina1,2,3,4
La fosfina, PH3, es el fosfuro de hidrógeno, un gas incoloro, inflamable y tóxico. La fosfina pura es inodora, pero normalmente contiene algunas impurezas, y entonces presenta un olor muy desagradable parecido al ajo o al pescado podrido, debido a la presencia de difosfina, P2H6, y de otras fosfinas sustituidas. Las fosfinas son también los compuestos organofosforados de fórmula PR3, donde R es un radical orgánico.
El primero en obtener fosfina fue Philippe Gengembre (1764-1838), un estudiante de Lavoisier, que la obtuvo en 1783 calentando fósforo en una solución acuosa de carbonato de potasio.
Inicialmente la fosfina estuvo tan asociada al fósforo elemental, que durante algún tiempo fue considerada una forma gaseosa del fósforo, hasta que, en 1789, Lavoisier indicó que era una combinación del fósforo con el hidrógeno, y la describió como fosfuro de hidrógeno.
En 1844, Paul Thénard, hijo del químico francés Louis Jacques Thénard, utilizó una trampa fría para separar la fosfina de la difosfina, que había obtenido a partir del fosfuro de calcio, y demostró con ello que la difosfina era la responsable de la inflamabilidad espontánea que normalmente presentaba la fosfina, y también del característico color naranja/marrón que se formaba en las superficies, consecuencia de su polimerización. Paul Thénard también consideró que la fórmula de la difosfina debía ser PH2 (en realidad es P2H4), intermedia entre el fósforo elemental y la fosfina.
La fosfina se emplea en la industria de los semiconductores y de los plásticos, en la producción de retardadores de llama, y como insecticida en locales cerrados, tales como silos de grano y viviendas.
Propiedades físico-químicas1,2,3,4,5
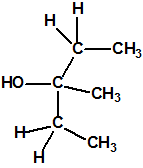
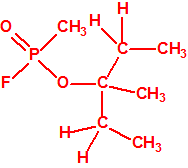
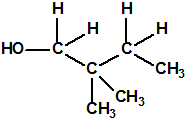
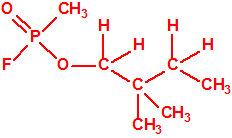
La fosfina, PH3, es el fosfuro de hidrógeno, también conocida como fosfuro de trihidrógeno, hidruro de fósforo, trihidruro de fósforo, Phosphorwasserstoff, etc. Su número CAS es 7803-51-2, su número EC es 232-260-8 y su número ONU es 2199. Es una molécula trigonal piramidal con simetría molecular C3V, longitud del enlace P-H de 1,42 Å, y ángulo del enlace H-P-H de 93,5°:
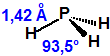
Es una molécula pequeña, de peso molecular 34,00 y densidad relativa de 1,18, punto de fusión de -132,8 °C, punto de ebullición de -87,7 °C, momento dipolar de 0,58 D y potencial de ionización de 9,96 eV.
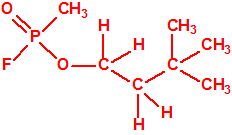
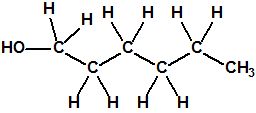
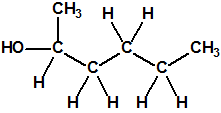
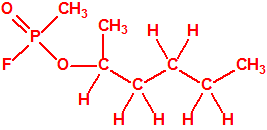
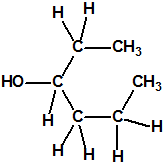
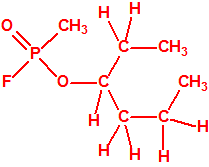
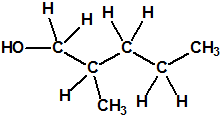
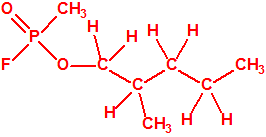
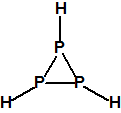
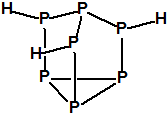
Obsérvese que el fósforo es el único elemento del grupo 15 (grupo del nitrógeno) que forma más de un hidruro. Además de la fosfina, PH3, forma entre otros, la difosfina, P2H4, CAS 13445-50-6, que normalmente se forma junto con la fosfina, pudiendo ambos separarse por condensación, debido a la diferencia de los puntos de ebullición (-87,7 °C frente a 63,5 °C), quedando la difosfina en forma de líquido amarillo. La difosfina se inflama espontáneamente en contacto con el aire, y se descompone con el tiempo formando polímeros sólidos amorfos, de color amarillo, insolubles en los disolventes orgánicos y de estequiometria variable, pero cercana a P2H. El fósforo también forma otros hidruros con diferentes estequiometrias, por ejemplo, PnH2n+2, PnHn, PnHn-2, PnHn-4, etc., siendo los más conocidos el trifosfano, P3H3, el pentafosfano, P5H5, y el heptafosfano, P7H3:
 |
 |
 |
| Trifosfano, ciclotrifosfano
CAS 20656-09-1 |
Pentafosfano, ciclopentafosfano
CAS 6798-45-4 |
Heptafosfano, trihidruro de heptafósforo
CAS 51273-53-1 |
La pureza de la fosfina influye sobre dos aspectos importantes de la misma, el olor y la inflamabilidad. Como ya se ha indicado la fosfina pura es inodora, pero la presencia de impurezas le confiere un olor desagradable que permite su detección en concentraciones del orden de 0,14-7 mg/m3. La temperatura de autoignición de la fosfina pura es de 38 °C pero la presencia de impurezas, en particular de difosfina, provoca con frecuencia que el producto técnico se inflame espontáneamente a temperatura ambiente, y forme mezclas explosivas con el aire en concentraciones superiores a 1,8% (LEL 1,79%).
La NFPA establece para la fosfina una clasificación de peligro:
| Peligro para la salud: 4 (extremo) |
 |
| Peligro de inflamabilidad: 4 (extremo) |
| Peligro por inestabilidad: 2 (moderado) |
La fosfina es escasamente soluble en agua, alrededor del 2,5 % (v/v) a 20 °C, pero es soluble entre un 2,5-15 % (v/v) en la mayor parte de los disolventes orgánicos a temperatura ambiente.
Las soluciones acuosas de fosfina no muestran propiedades ácidas ni básicas. Las investigaciones sobre el intercambio de deuterio entre el agua deuterada y la fosfina revelaron que este intercambio procede vía un ión PH4+ en soluciones ácidas y a través de un ión PH2– en soluciones básicas. A partir de los datos cinéticos y de los mecanismos de intercambio, se establecieron los siguientes valores para las constantes de equilibrio, a 27 °C:
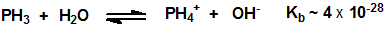

Como muchos hidruros la fosfina es un poderoso agente reductor:
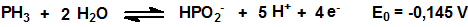
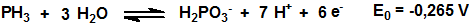
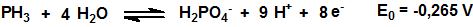
El poder reductor de la fosfina se utiliza, por ejemplo, para su detección mediante el empleo de tubos colorimétricos.
La oxidación de la fosfina produce agua y óxidos u oxiácidos de fósforo:

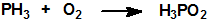

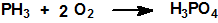
La fosfina en atmósfera de cloro elemental arde con formación de pentacloruro de fósforo, PCl5, y cloruro de hidrógeno, HCl. Las soluciones acuosas de cloro oxidan la fosfina a ácido fosfórico:


La fosfina reacciona casi instantáneamente con las soluciones de hipoclorito de sodio, por lo que estas se emplean para atrapar las trazas de fosfina en una corriente gaseosa:

Obtención de la fosfina1,6
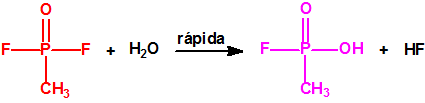
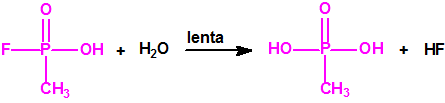
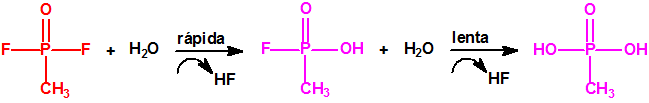
A escala de laboratorio la fosfina se prepara fácilmente mediante la hidrólisis de alguno de sus fosfuros metálicos, por ejemplo, del fosfuro de aluminio, AlP, CAS 20859-73-8, del fosfuro de calcio, Ca3P2, CAS 1305-99-3, del fosfuro de magnesio, Mg3P2, CAS 12057-74-8, o del fosfuro de zinc, Zn3P2, CAS 1314-84-7. Para la hidrólisis, además del agua, pueden utilizarse ácidos o bases, y mezclas acuosas de ácidos o bases con solventes orgánicos (como, por ejemplo, dioxano o alcoholes). En la hidrólisis del fosfuro de calcio, además de fosfina se forman difosfina y otros hidruros de fósforo, y si el fosfuro de calcio se ha obtenido por reducción del Ca3(PO4)2 con carbón, en su hidrólisis puede aparecer hasta un 3% de acetileno. Todo indica que el mejor método de laboratorio es la hidrólisis del fosfuro de aluminio, de la mayor pureza posible, con agua fría, para así evitar la formación de difosfina y otras impurezas, responsables de la inflamabilidad espontánea.


También puede obtenerse mediante reacción del fósforo con el hidrógeno:

Los dos métodos industriales más comunes se basan en reacciones de desproporción del fósforo elemental catalizadas por álcalis o ácidos. Por ejemplo, el fósforo blanco reacciona con hidróxido sódico para producir fosfina e hipofosfito de sodio, NaH2PO2, CAS 7681-53-0:
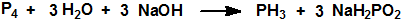
En las plantas de producción de hipofosfito de sodio, la fosfina es un subproducto que generalmente se quema. En presencia de alcoholes, se puede controlar la reacción de modo que se produzca fosfina y fosfito de disódico Na2HPO3, CAS 13708-85-5, con un mejor rendimiento:

En la reacción de desproporción catalizada por ácidos, a 280 °C, el fósforo blanco se convierte en fosfina y ácido fosfórico, con la formación de fósforo rojo como producto intermedio. La reacción tiene un buen rendimiento pero debido a la alta temperatura y a la corrosividad del medio, requiere un reactor de grafito:

Detección de la fosfina7,8,9,10
Existen tubos colorimétricos para la determinación semi-cuantitativa de fosfina en diversos rangos de concentración, que van desde 0,1-4 ppm hasta 200-10000 ppm, de diferentes fabricantes, con diferentes reacciones, diferente número de emboladas y diferentes interferencias.
Algunos tubos emplean el poder reductor de la fosfina para detectar su presencia mediante la reducción del ión Au3+ con formación de oro metálico coloidal, produciéndose un cambio de color de amarillo a marrón oscuro o gris-violeta:

Otros hidruros como la arsina, AsH3, y la estibina, SbH3, también dan la misma reacción pero con diferente sensibilidad.
Otros tubos emplean la formación de un complejo con cloruro mercúrico que provoca la formación de ácido clorhídrico y un cambio de color en el indicador de pH:

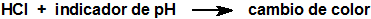
También son muy empleados por su simplicidad y economía los sensores o monitores personales que emplean sensores electroquímicos. Los sensores electroquímicos pueden emplearse en sistemas detectores individuales o en sistemas de detección multi-gases, trabajan en el rango de 0,1-5 ppm, con una resolución de 0,1 ppm y un tiempo de respuesta del orden de 60 segundos.
Estos sensores, de poco peso y fácil manejo, suelen colocarse colgados del cuello o sujetos a la ropa cerca de nariz, y suelen disponer de dos alarmas; una suena cuando detecta una concentración de 0,3 ppm de fosfina y la otra suena cuando se alcanza una concentración mayor, por ejemplo, 0,6 ppm.
La fosfina con un potencial de ionización de 9,96 eV puede detectarse mediante el empleo de un detector de fotoionización (PID, PhotoIonization Detector). Según indica el fabricante es posible emplear cualquiera de las tres lámparas más habituales (de 9,8 eV, de 10,6 eV y de 11,7 eV). La más empleada es la de 10,6 eV por su buena sensibilidad y duración. En concentraciones muy altas, si existen interferencias cruzadas, puede ser útil la lámpara de 9,8 eV. La principal ventaja de los detectores de fotoionización sobre los tubos colorimétricos y los sensores electroquímicos es su mejor tiempo de respuesta, de segundos, frente a minutos. Además el sistema de bombeo de los PID permite el empleo de sondas y la comprobación de fugas en contenedores y en recipientes estancos.
No todo son ventajas, en el caso de la fosfina, los PID sufren el fenómeno de empañamiento de la lámpara, «Lamp Fogging». La fosfina es una de las pocas sustancias que reacciona foto-químicamente y forma sustancias que empañan la superficie de la lámpara del PID. Esto ocurre con todas las lámparas, y el efecto es tanto mayor, cuanto mayor es la concentración de fosfina y mayor el tiempo de exposición a la misma. El efecto es más que evidente a concentraciones de fosfina unos cientos de ppm, pero incluso a unos 20 ppm se nota, en cuestión de minutos, una disminución sensible de la respuesta. Exposiciones cortas e intermitentes ayudan a minimizar la acumulación de dichos recubrimientos, que se quitan fácilmente limpiando el cristal de la lámpara con metanol anhidro.
La fotometría de llama, que detecta y mide la emisión característica de diferentes elementos químicos, entre ellos el fósforo, se emplea tanto en los equipos de campo como en los equipos de laboratorio para la detección y cuantificación de la fosfina. Los detectores portátiles de fotometría de llama, AP2C y AP4C permiten la detección de fósforo, y por tanto de fosfina, difosfina y otros hidruros de fósforo, con una excelente sensibilidad, pero con escasa selectividad, ya que la respuesta es de tipo «atómico».

Detector fotometrico de llama, AP2C, mostrando en rojo la señal de detección en la línea del fósforo, debido a la presencia de fosfina
La fosfina como fumigante11,12,13,14,15,16,17
La fumigación con fosfina se realiza lleva a cabo sobre productos almacenados en cámaras, almacenes, silos, almacenes de alimentos, contenedores, vagones de tren, barcazas, buques, aviones, etc., y también se lleva a cabo como medida de desinsectación o de cuarentena contra plagas en estructuras tales como molinos de harina o fábricas de alimentos, o en contenedores vacíos y otros medios de transporte. Para conseguir una buena eliminación de las plagas, en todas las etapas de su ciclo de vida (huevo, larva, pupa, adulto), se requiere alcanzar y mantener una concentración de fosfina de 300 ppm durante siete días (con una temperatura de 25 °C) o de 200 ppm durante 10 días (con una temperatura de 15-25 °C), debiendo comprobarse la concentración mediante algún sistema de detección. Además para una fumigación eficiente y segura se requiere que la zona a fumigar sea estanca. Por ejemplo, el gobierno australiano ha establecido, en la norma Australian Standard 2628 (AS2628), la estanqueidad que deben tener los silos. La norma requiere que para considerar el silo estanco la sobrepresión debe tardar más de cinco minutos en descender de 25 mm a 12,5 mm.
Las preparaciones comerciales para fumigación contienen, en general, como ingrediente activo, alrededor de un 57% de fosfuro de aluminio, o alrededor de un 34% de difosfuro trimagnésico, que en contacto con la humedad del ambiente generan lentamente fosfina, durante su hidrólisis:
Phostoxin (contiene un 55 % de fosfuro de aluminio) es una preparación comercial sólida, en forma de pellets o bolas, que lentamente libera fosfina en contacto con la humedad en el aire. Existen muchas otras preparaciones comerciales sólidas muy similares, como por ejemplo, Celphos, Quickphos, Gastowin, Delitia, Detia gas, Magtoxin, Agtoxin, etc.. Después del tratamiento con fosfina, las zonas deben ventilarse, y comprobar que los niveles de fosfina son los permitidos para que los trabajadores puedan entrar con seguridad a los recintos fumigados.
Algunas preparaciones contienen aditivos, como por ejemplo, carbamato de amonio NH2COONH4, CAS 1111-78-0, ingrediente inerte aprobado para su uso en las formulaciones con fosfuro de aluminio, utilizado para reducir el riesgo de incendio de la fosfina. Para suprimir la inflamabilidad (autoinflamación) de la fosfina, el carbamato de amonio libera amoníaco y dióxido de carbono que diluye la fosfina formada en la reacción de hidrólisis. El amoníaco sirve también como un agente de advertencia. Reassessmen inerte-amonio carbamato EPA.
Para la fumigación se emplean diferentes métodos de aplicación entre los que se incluyen: tratamiento en superficie (Surface application), sonda (probing, sub-surface treatment, trench-in), utilización de un tubo perforado colocado en el fondo (perforated tubing laid at the bottom of spaces), y sistemas de recirculación y de inyección de gas (recirculation systems and gas-injection systems), entre otros. La periodicidad del tratamiento dependerá de la temperatura, la profundidad de la carga y el método utilizado.
Al realizar la fumigación se deberán colocar señales de advertencia en todas las entradas a los lugares fumigados y en aquellos espacios que se consideren peligrosos durante la fumigación. En las señales se incluirán los datos de la fumigación y la fecha y hora en que se llevó a cabo.
Se debe garantizar que todos los desechos y residuos de activos que pudieran generar fosfina, se eliminan del modo apropiado, ya que suponen un peligro para las personas que pudieran entrar en contacto con ellos.
Toxicidad de la fosfina18,19,20,21,22,23,24,25
La fosfina es muy tóxica. La máxima concentración de fosfina en el ambiente no debe exceder de 0,3 ppm para una jornada laboral de 8 horas y una semana laboral de 40 horas (el TLV como TWA es 0,3 ppm ó 0,4 mg/m3).
Para la fosfina, el valor TLV-TWA (Threshold Limit Value – Time Weighted Average), valor límite umbral para un valor medio de exposición de una jornada laboral de ocho horas y una semana laboral de 40 horas, es de 0,3 ppm.
Para la fosfina, el valor TLV-STEL (Threshold Limit Value – Short Term Exposure Limit), valor límite umbral para una exposición a corto plazo, o concentración máxima para un límite de exposición continua de 15 minutos (con un máximo de cuatro periodos por día con al menos 60 minutos entre periodos de exposición) es de 1ppm.
En decir, los trabajadores no deben ser expuestos más de cuatro veces por día a más de 1ppm durante más de 15 minutos, con al menos una hora entre cada exposición. Y los trabajadores no deben estar expuestos a más de 0,3 ppm durante más de ocho horas diarias o 40 horas a la semana.
El olor de la fosfina es perceptible cuando su concentración alcanza o excede las 2 ppm, un valor mucho mayor que su TLV. Esto significa que cuando el trabajador percibe el olor a fosfina la concentración de ésta supera los límites de exposición segura.
Una concentración de 50-100 ppm puede ser soportada sin daño sólo por un tiempo muy corto, y una concentración de 400 ppm conduce rápidamente a la muerte. Los síntomas observados para los envenenamientos moderados y graves por fosfina son: sensación de ansiedad, sensación de presión en el pecho, falta de aire, dolor detrás del esternón, tos seca ocasional, incremento del ruido al respirar, confusión, vértigo y desvanecimiento. Como primeros auxilios, aparte a la víctima a una zona de aire limpio y si es posible suminístrele oxígeno.
El valor revisado para el IDLH de la fosfina es 50 ppm, aunque este valor puede ser un valor conservador debido a la falta de datos relevantes de toxicidad aguda para los trabajadores expuestos a concentraciones superiores a 35 ppm. https://www.cdc.gov/niosh/idlh/7803512.html
En caso de un accidente con fosfina, la «Guía de Respuesta en caso de Emergencia», GRE2016, aconseja el empleo de la Guía nº 119, «Gases – Tóxicos – Inflamables», cuyo contenido se reproduce a continuación:
PELIGROS POTENCIALES
A LA SALUD
- TÓXICO; puede ser fatal si se inhala o se absorbe por la piel.
- El contacto con gas o gas licuado puede causar quemaduras, lesiones severas y/o quemaduras por congelación.
- El fuego producirá gases irritantes, corrosivos y/o tóxicos.
- Las fugas resultantes del control del incendio pueden causar contaminación.
INCENDIO O EXPLOSION
- Inflamable; puede encenderse por calor, chispas o llamas.
- Puede formar mezclas explosivas con el aire.
- Aquellas sustancias designadas con una (P) pueden polimerizar explosivamente cuando se calientan o están involucradas en un incendio.
- Los vapores de gas licuado son inicialmente más pesados que el aire y se esparcen a través del piso.
- Los vapores pueden viajar a una fuente de encendido y regresar en llamas.
- Algunos de estos materiales pueden reaccionar violentamente con agua.
- Los cilindros expuestos al fuego pueden ventear y liberar gases tóxicos e inflamables a través de los dispositivos de alivio de presión.
- Los contenedores pueden explotar cuando se calientan.
- Los cilindros con rupturas pueden proyectarse.
- La fuga resultante del control puede crear incendio o peligro de explosión.
SEGURIDAD PUBLICA
- LLAMAR primero al número de teléfono de respuesta en caso de emergencia en el documento de embarque. Si el documento de embarque no está disponible o no hay respuesta, diríjase a los números telefónicos enlistados en el forro de la contraportada.
- Cómo acción inmediata de precaución, aisle el área del derrame o escape como mínimo 100 metros (330 pies) en todas las direcciones.
- Mantener alejado al personal no autorizado.
- Manténgase con viento a favor, en zonas altas y/o corriente arriba.
- Muchos de los gases son más pesados que el aire y se dispersan a lo largo del suelo y se juntan en las áreas bajas o confinadas (alcantarillas, sótanos, tanques).
- Ventile los espacios cerrados antes de entrar.
ROPA PROTECTORA
- Use el equipo de aire autónomo de presión positiva (SCBA).
- Use ropa protectora contra los productos químicos, la cual esté específicamente recomendada por el fabricante. Esta puede proporcionar poca o ninguna protección térmica.
- El traje de protección estructural de los bomberos provee protección limitada ÚNICAMENTE en situaciones de incendio; no es efectivo en derrames con posible contacto directo con la sustancia.
EVACUACIÓN
Derrame
- Vea la Tabla 1 – Distancias de Aislamiento Inicial y Acción Protectora para los materiales resaltados. Para los otros materiales, aumente como sea necesario en la dirección del viento, la distancia de aislamiento mostrada en «SEGURIDAD PUBLICA».
Incendio
- Si un tanque, carro de ferrocarril o autotanque está involucrado en un incendio, AISLE a la redonda a 1600 metros (1 milla) también, considere la evacuación inicial a la redonda a 1600 metros (1 milla).
En Canadá, puede requerirse para este producto un Plan de Asistencia en Respuesta a Emergencias (ERAP). Por favor consulte los documentos de embarque y/o la sección Programa ERAP.
RESPUESTA DE EMERGENCIA
FUEGO
- NO EXTINGA UN INCENDIO DE FUGA DE GAS A MENOS QUE LA FUGA PUEDA SER DETENIDA.
Incendio Pequeño
- Polvos químicos secos, CO2, rocío de agua o espuma resistente al alcohol.
Incendio Grande
- Use rocío de agua, niebla o espuma resistente al alcohol.
- PARA CLOROSILANOS, NO USE AGUA, use espuma AFFF resistente al alcohol como medio de expansión.
- Mueva los contenedores del área de fuego si lo puede hacer sin ningún riesgo.
- Los cilindros dañados, deberán ser manejados solamente por especialistas.
Incendio que involucra Tanques
- Combata el incendio desde una distancia máxima o utilice soportes fijos para mangueras o chiflones reguladores.
- Enfríe los contenedores con chorros de agua hasta mucho después de que el fuego se haya extinguido.
- No ponga agua directamente a la fuente de la fuga o mecanismos de seguridad; puede ocurrir congelamiento.
- Retírese inmediatamente si sale un sonido creciente de los mecanismos de seguridad de las ventilas, o si el tanque se empieza a decolorar.
- SIEMPRE manténgase alejado de tanques envueltos en fuego.
DERRAME O FUGA
- ELIMINAR todas las fuentes de ignición (no fumar, no usar bengalas, chispas o llamas en el área de peligro).
- Todo el equipo que se use durante el manejo del producto, deberá estar conectado eléctricamente a tierra.
- Deberán usarse trajes protectores de encapsulamiento total contra el vapor, en derrames y fugas sin fuego.
- No tocar ni caminar sobre el material derramado.
- Detenga la fuga, en caso de poder hacerlo sin riesgo.
- No ponga agua directamente al derrame o fuente de la fuga.
- Use rocío de agua para reducir los vapores; o desviar la nube de vapor a la deriva. Evite que flujos de agua entren en contacto con el material derramado.
- PARA CLOROSILANOS, use espuma AFFF resistente al alcohol como medio de expansión para reducir los vapores.
- Si es posible, voltee los contenedores que presenten fugas para que escapen los gases en lugar del líquido.
- Prevenga la entrada hacia vías navegables, alcantarillas, sótanos o áreas confinadas.
- Aisle el área hasta que el gas se haya dispersado.
PRIMEROS AUXILIOS
- Asegúrese que el personal médico tenga conocimiento de los materiales involucrados, y tomar las precauciones para protegerse a sí mismos.
- Mueva a la víctima a donde se respire aire fresco.
- Llamar a los servicios médicos de emergencia.
- Aplicar respiración artificial si la víctima no respira.
- No usar el método de respiración de boca a boca si la víctima ingirió o inhaló la sustancia: proporcione la respiración artificial con la ayuda de una máscara de bolsillo con una válvula de una sola vía u otro dispositivo médico de respiración.
- Suministrar oxígeno si respira con dificultad.
- Quitar y aislar la ropa y el calzado contaminados.
- En caso de contacto con la sustancia, enjuagar inmediatamente la piel o los ojos con agua corriente por lo menos durante 20 minutos.
- En caso de contacto con gas licuado, descongelar las partes con agua tibia.
- En caso de quemaduras, inmediatamente enfríe la piel afectada todo el tiempo que pueda con agua fría. No remueva la ropa que está adherida a la piel.
- Mantenga a la víctima calmada y abrigada.
- Mantener a la víctima bajo observación.

De acuerdo al GRE2016, las distancias de aislamiento y de protección para la fosfina y algunos fosfuros (en el caso de que estos últimos entren en contacto con el agua) son las siguientes:
| Nº Guía |
Nº ONU |
Nombre |
Derrames pequeños |
Derrames grandes |
| Daislam |
Dprotec |
Daislam |
Dprotec |
| Día |
Noche |
Día |
Noche |
| 119 |
2199 |
Fosfina |
60 m |
200 m |
1000 m |
300 m |
1,3 km |
3,8 km |
| 139 |
1397 |
Fosfuro de aluminio (cuando es derramado en el agua) |
60 m |
200 m |
900 m |
500 m |
2 km |
7,1 km |
| 139 |
1360 |
Fosfuro de calcio (cuando es derramado en el agua) |
30 m |
200 m |
600 m |
300 m |
1 km |
3,7 km |
| 139 |
2011 |
Fosfuro de magnesio (cuando es derramado en el agua) |
60 m |
200 m |
800 m |
400 m |
1,7 km |
5,7 km |
| 157 |
3048 |
Plaguicida a base de fosfuro de aluminio (cuando es derramado en el agua) |
60 m |
200 m |
900 m |
500 m |
2 km |
7 km |
Es muy importante respetar todas las indicaciones, recomendaciones, consejos y advertencias que se incluyen en las etiquetas y documentos informativos de los productos utilizados para fumigación, a fin de evitar cualquier tipo de incidente mortal como los que por desgracia se producen todos los años.26,27,28,29,30,31
Referencias
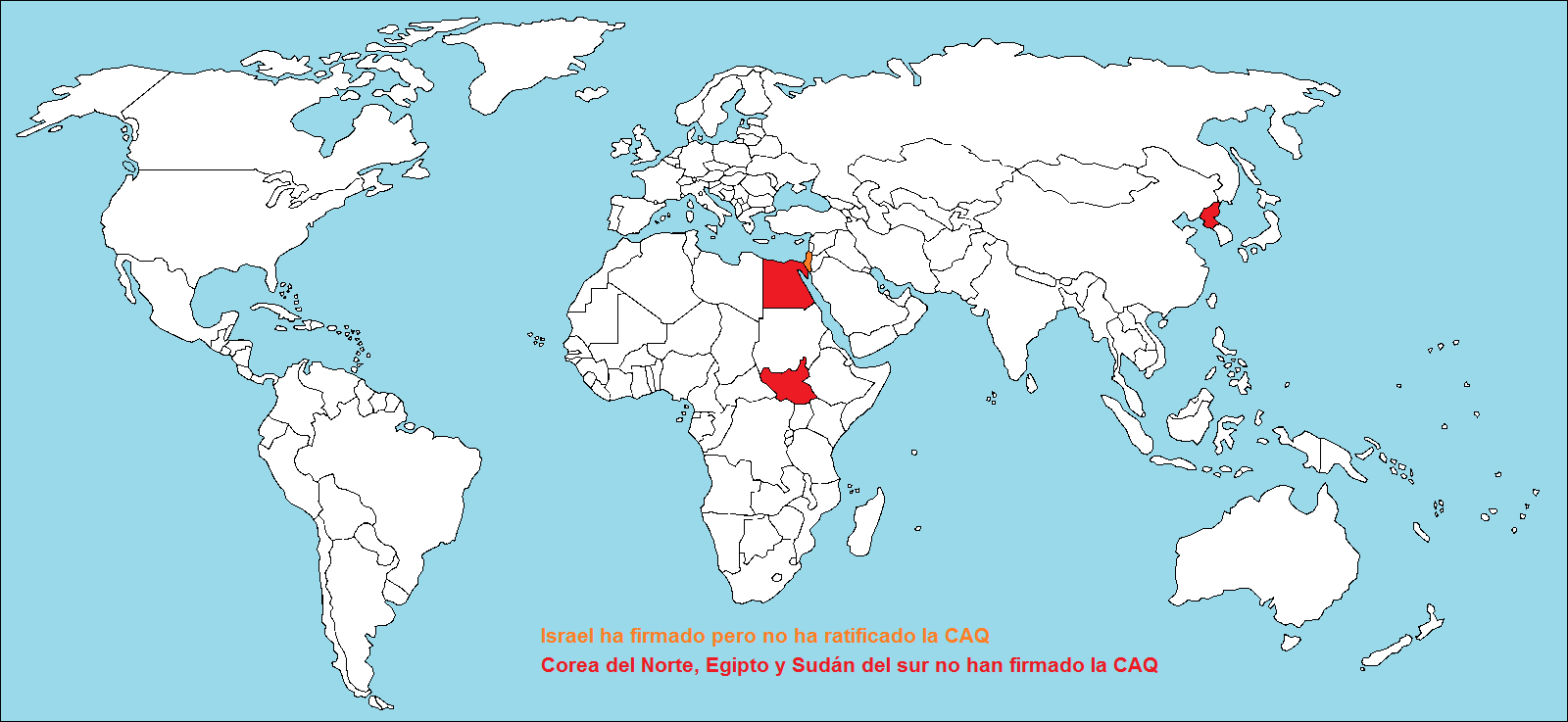
- «Phosphine» , https://en.wikipedia.org/wiki/Phosphine
- «Phosphine», INCHEM, http://www.inchem.org/documents/pims/chemical/pim865.htm
- «The chemistry of phosphine», Ekkehard Fluck, https://www.thevespiary.org/rhodium/Rhodium/Vespiary/talk/files/2494-The-Chemistry-of-Phosphine807f.pdf
- «Phosphine and selected metal phosphides», Environmental Health Criteria 73, International Programme on Chemical Safety, http://www.inchem.org/documents/ehc/ehc/ehc73.htm
- «Atlas d´équilibres électrochimiques à 25 °C», Marcel Pourbaix, Gauthier-Villars & Cie, 1963
- «Ullmann’s Encyclopedia of Industrial Chemistry», «Phosphorus Compounds, Inorganic», 7th Ed.
- «Measurement of fumigants in the food storage industry», RAE Systems, Application Note 218, http://www.raesystems.com/sites/default/files/content/resources/Application-Note-218_Measurement-Of-Fumigants-In-The-Food-Storage-Industry_08-05.pdf
- «Dräger-Tubes & CMS-Handbook», Dräger, 2011, 16th., https://www.google.es/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=0ahUKEwjiitnVnJ3UAhUFSRoKHTa8DpwQFggsMAA&url=https%3A%2F%2Fwww.draeger.com%2FLibrary%2FContent%2Ftubeshandbook_br_9092086_en.pdf&usg=AFQjCNF3QlaZZEy40nmdZjJE2AErhabXCA
- «Gas Detection Tubes and Sampling Handbook», RAE, 2ªEd., 2013, http://www.raesystems.com/sites/default/files/content/resources/eBook-gas-detection-tube-and-sampling-handbook.pdf
- «The PID handbook», RAE, 3ªEd., 2013, http://www.raesystems.com/sites/default/files/content/resources/pid_handbook_1002-02.pdf
- «Fumigation in the 21st century», C.H. Bell, Crop Protection 19 (2000) 563-569
- «Fumigating with phosphine, other fumigants and controlled atmospheres», Grains Research & Development Corporation, http://storedgrain.com.au/wp-content/uploads/2016/10/GRDC-PHOSP-Booklet_2016_R2_Reduced.pdf
- «Phostoxin-Mg Placas», Detia Freyberg GmbH, http://www.grupoavisur.com/docs/MADERA/04_AVISUR_MADERA_PHOSTOXIN_FICHA_SEGURIDAD.pdf
- «Fosfuro de aluminio-KILLPHOS», FAX, http://www.faxsa.com.mx/Fosf_MT/KillPhMT.pdf
- «Quickphos Fumigation Tablets and Pellets», http://www.fumigationservice.com/pdf/MSDS_ALP.pdf
- «Inert Reassessment-Ammonium Carbamate», EPA, https://www.epa.gov/sites/production/files/2015-04/documents/carbamate.pdf
- «Carga y estiba en buques BULK CARRIER», A. Barrera Acosta & M. Carbonell Casadesús, Universidad de La Laguna, https://www.google.es/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=0ahUKEwiyrry8za7UAhXHLFAKHb-DBkAQFggpMAA&url=https%3A%2F%2Friull.ull.es%2Fxmlui%2Fbitstream%2Fhandle%2F915%2F4365%2FCARGA%2520Y%2520ESTIBA%2520EN%2520BUQUES%2520BULK%2520CARRIER.pdf%3Fsequence%3D1&usg=AFQjCNFRSB1_l57ek_9rUiNTZBqcstLgwg
- «Phosphine», CDC, https://www.cdc.gov/niosh/idlh/7803512.html
- «Phosphine», NIOSH, https://www.cdc.gov/niosh/docs/2003-154/pdfs/6002.pdf
- «Code of practice phosphine», Asia Industrial Gases Association, IGC Doc 162/10/E, http://eiga.web1.apollo-com.be/fileadmin/docs_pubs/Doc_162_10_E.pdf
- «FISQ Fosfuro de aluminio», INSHT, http://www.insht.es/InshtWeb/Contenidos/Documentacion/FichasTecnicas/FISQ/Ficheros/401a500/nspn0472.pdf
- «FISQ Fosfuro de calcio», INSHT, http://www.insht.es/InshtWeb/Contenidos/Documentacion/FichasTecnicas/FISQ/Ficheros/1101a1200/nspn1126.pdf
- «FISQ Fosfuro de magnesio», INSHT, http://www.insht.es/InshtWeb/Contenidos/Documentacion/FichasTecnicas/FISQ/Ficheros/701a800/nspn0744.pdf
- «FISQ Fosfuro de zinc», INSHT, http://www.insht.es/InshtWeb/Contenidos/Documentacion/FichasTecnicas/FISQ/Ficheros/601a700/nspn0602.pdf
- «Guía de Respuesta en caso de Emergencia», GRE2016, https://www.tc.gc.ca/media/documents/tmd-fra/SpanishERGPDF.pdf
- «Cuatro menores mueren en Texas tras inhalar el gas tóxico de un pesticida que su familia utilizó para matar ratas», http://www.univision.com/dallas/kuvn/noticias/muertes/cuatro-menores-mueren-en-texas-tras-inhalar-el-gas-toxico-de-un-pesticida-que-su-familia-utilizo-para-matar-ratas
- «Muere un matrimonio en Burgos por una intoxicación con fitosanitarios contra polillas», http://www.abc.es/espana/castilla-leon/abci-fallece-matrimonio-burgos-intoxicacion-fitosanitarios-contra-polillas-201705161859_noticia.html
- «Texas pesticide deaths-Chemical may have sickened, but cleanup was fatal», http://edition.cnn.com/2017/01/03/health/texas-pesticide-deaths/
- «The Phosphine Accident – A Tragic Chemistry Lesson», http://www.acsh.org/news/2017/01/05/phosphine-accident-%E2%80%93-tragic-chemistry-lesson-10680
- «Un matrimonio burgalés, última víctima del fosfuro de aluminio», http://www.elespanol.com/ciencia/salud/20170516/216478876_0.html
- «Beware The Fiery Fumigator», http://maritimeaccident.org/2015/04/beware-the-fiery-fumigator/#more-21548